
” Neuropatia Uditiva e Dis-Sincronia Uditiva” (Auditory Neuropathy/ Auditory Dis-sinchrony: AN/AD- Auditory Neuropathy Spectrum Disorder (ANSD)
In questa pagina:
· Che cosa è la neuropatia uditiva Dis-Sincronia Uditiva?
· Quali sono le cause neuropatia Uditiva Dis-Sincronia uditiva?
· Quali sono i ruoli delle cellule ciliate esterne ed interne?
· Ci sono fattori di rischio per la neuropatia uditiva Dis-Sincronia Uditiva?
· Come viene diagnosticata la neuropatia uditiva Dis-Sincronia Uditiva?
· Ha neuropatia uditiva mai stare meglio o peggio?
Che cosa è la neuropatia uditiva e Dis-Sincronia Uditiva?
I termini ” neuropatia uditiva e dis-sincronia uditiva” (Auditory Neuropathy/ Auditory Dis-sinchrony: AN/AD) descrivono una forma di disordine uditivo di incerta classificazione, ma sufficientemente caratterizzata per quanto riguarda i dati forniti dall’ audiologia clinica. Infatti rientrano attualmente nella definizione i casi in cui è possibile verificare strumentalmente una normale funzione delle cellule cigliate esterne ed una concomitante disfunzione della via uditivo afferente, La Neuropatia uditiva è un disturbo dell’udito in cui il suono passa normalmente dall’orecchio medio all’orecchio interno, ma la trasmissione dei segnali dall’orecchio interno alle vie uditive centrali e corteccia uditiva è compromessa. Questa condizione è stata descritta più spesso nell’infanzia, e si ritiene che potrebbe interessare circa il 7-10% dei bambini con ipoacusie permanenti. Anche gli adulti possono manifestare questa forma, ma la prevalenza non è nota. Le persone con Neuropatia Uditiva possono avere un udito normale oppure la perdita dell’udito può variare da lieve a grave; hanno sempre scarse abilità vocale di percezione, nel senso che hanno difficoltà a capire chiaramente il discorso. L’ AN/AD è clinicamente definita dalla presenza di otoemissioni, dall’assenza di risposte evocate troncoencefaliche (ABP), dall’assenza di riflessi stapediali, da una perdita uditiva di entità variabile, e da una soglia di percezione verbale innalzata peggiore rispetto a quanto sarebbe previsto in base al soglia uditiva tonale (grado di perdita uditiva).
Quali sono le cause della neuropatia uditiva?
L’ampia variabilità delle manifestazioni cliniche della AN/AD potrebbe riflettere vari stadi della stessa patologia oppure potrebbe essere l’espressione di disordini nelle diverse sedi della via uditiva. Queste comprendono le cellule cigliate interne, le sinapsi fra cellule cigliate interne e fibre afferenti del nervo cocleare, e le stesse fibre del nervo. Anche se le cellule ciliate cellule ciliate esterne adiacenti alle vie acustiche sono più numerose rispetto alle cellule ciliate interne, le esterne sono generalmente più soggette a danni rispetto alle cellule ciliate interne, tuttavia le cellule ciliate esterne sembrano funzionare normalmente in persone con neuropatia uditiva. La Neuropatia Uditiva (AN) è un disturbo della funzionalità uditiva che si ritiene determini danni alle cellule ciliate interne, e connessioni difettose tra le cellule ciliate interne e il nervo che porta informazioni dall’orecchio interno al cervello, o danni al nervo stesso. La Figura 1 mostra il confronto tra i nervi uditivi tra due persone con funzione uditiva normale e con AN. Vi è una notevole riduzione del numero di fibre nervose uditive nel paziente AN. Il nervo surale nel paziente ha mostrato anche la perdita assonale con evidenza di demielinizzazione e rimielinizzazione. La figura 3 mostra le sezioni dei nervi surale da soggetto udito e normale come da un soggetto. Si può vedere chiaramente la perdita di grandi fibre mieliniche nel soggetto con AN e l’assottigliamento della mielina in molti delle grandi fibre rimanenti

Figura 1 microfotografie di una sezione trasversale del nervo uditivo in un paziente con neuropatia sensoriale-motoria ereditaria e un’altra persona con udito normale

Figura 2 Sezione trasversale di un normale biopsia del nervo surale e un nervo surale da un soggetto con neurapatia uditiva con una concomitante neuropatia periferica
Sono stati proposti i seguenti anomalie:
• Lesioni alla giunzione sinaptica tra le cellule ciliate interne della coclea e dendriti dei neuroni gangliari a spirale
• Danno diretto alle dendriti dei neuroni gangliari spirale
• Danno diretto ai neuroni gangliari spirale
• Danno diretto assonale al nervo uditivo che provoca una cascata di danni al nuclei uditivo più bassa
APPROFONDIMENTO emc
La presenza di segnali normali provenienti dalle cellule ciliate esterne della coclea (otoemissioni e potenziali microfonici) segna la normalità dei processi micromeccanici di trattamento sonoro (amplificazione in filtraggio), ma l’assenza di PEU precoci, come anche la loro onda I, è indicativa di un’anomalia della conduzione neurale che si manifesta fin dal ganglio spirale, o fin dalla sinapsi tra le cellule ciliate interne e i neuroni del nervo cocleare. L’assenza d’onda nei PEU precoci non implica assolutamente l’assenza di percezione, e d’altronde le neuropatie uditive si caratterizzano anche da una discordanza tra PEU assenti e audiometria tonale preliminare soggettiva non necessariamente molto anormale. L’interpretazione fisiologica delle neuropatie uditive è che i potenziali di azione sono mal sincronizzati, e ciò soprattutto a causa di un deficit dei neuroni del ganglio spirale: la presenza di potenziali d’azione permette l’individuazione dei suoni di base ma il cattivo cadenzamento temporale rende non evidenziabile l’attività elettroencefalografica associata. L’identificazione dei suoni complessi (parola, parola nel rumore), che richiede indici temporali precisi, è degradata dalla dissincronia delle scariche neuronali: questa caratteristica si verifica facilmente in audiometria. Le stime di prevalenza dell’entità NU-DU nella popolazione affetta da deficienza uditiva confermata oscillano tra il 4% e l’11%. Questa spiegazione per difetto della sincronizzazione neuronale non è tuttavia la sola causa plausibile. Per questo, la denominazione attualmente ritenuta è meno esplicita sul fenomeno fisiopatologico. Parlando di neuropatia uditiva con disordini di spettro (NA-DS), si rimane descrittivi, e conseguentemente non si rigetta la possibilità di neuropatie senza dissincronia.
L’evidenziazione di un interessamento già a livello del nervo uditivo (e a volte anche, per estensione, a livello presinaptico nelle cellule ciliate interne come nella mutazione dell’otoferlina), evidenziata dall’assenza d’onda I nei PEUTC, fa sì che le NA-DS non siano a priori classificate nei deficit uditivi centrali. Tuttavia, è stato dimostrato che alcune neuropatie uditive ledono in maniera diffusa le vie uditive, non solamente il nervo uditivo ma anche le vie più centrali, il che lascia presagire conseguenze percettive specifiche in relazione con le funzioni proprie ai neuroni centrali deficitari. Recentemente, il gene che codifica per una molecola chiamata pejvakin, insufficiente nella sordità DFNB59, è stato identificato e sequenziato a partire da uno studio genetico di quattro famiglie consanguinee che vivono in isolati geografici [71]. I soggetti colpiti presentano tutti una sordità neurosensoriale prelinguale bilaterale isolata, e le mutazioni omozigote del gene della pejvakin che sono state identificate sono due e tutte e due di tipo falso-senso. La sordità DFNB59 presenta un profilo laterale molto particolare, con un interessamento audiometrico a plateau che può essere solo severo, otoemissioni presenti e PEU molto degradati con, in alcuni pazienti, un’onda V singola riconoscibile, di latenza allungata. La funzione della pejvakin è al momento ancora sconosciuta. Un topo transgenico (knock in) è stato creato con la principale mutazione identificata del gene della pejvakin; presenta una sordità più moderata che nell’uomo, che non supera i 40 dB e spesso limitata alle alte frequenze. Le otoemissioni sono rigorosamente normali, ma i PEUTC presentano anomalie nette della conduzione, con un allungamento marcato degli intervalli interonde I-II, II-III e III-IV, il che segna la natura mista, centrale e periferica, della lesione neurale. La proteina peraltro presente lungo tutte le vie nervose uditive fino al collicolo inferiore (come d’altra parte nelle cellule ciliate esterne della coclea, dove la forma con la mutazione falso-senso non sembra indurre disfunzione), ha chiaramente un ruolo nei neuroni uditivi centrali.
Altre famiglie sono state identificate a seguito dell’osservazione principe, anche queste portatrici di mutazioni della pejvakin. In una di loro, in Marocco, il deficit uditivo è evolutivo e gli autori lo attribuiscono a un’origine strettamente cocleare (OEA assenti, PEU non registrabili, assenza di potenziali microfonici) [72]. Stranamente, gli autori non rilevano che malgrado una sordità a plateau limitata a 70 dB in alcuni pazienti, questi non hanno PEU anche a 105 dB e la loro protesi uditiva è risultata inefficace (l’audiometria vocale con protesi era scarsa). Questo indica pertanto la presenza di una componente neuropatica periferica e forse centrale, come nelle famiglie iraniane. In ogni caso, questo esempio illustra l’eterogeneità clinica che può essere riscontrata in occasione di mutazioni diverse di uno stesso gene, e l’eterogeneità delle osservazioni cliniche che, quando sono incompletamente analizzate, misconoscono a volte la componente centrale di una sordità mista, periferica e centrale.
Fino a qualche anno fa non era mai stata osservata una lesione uditiva centrale specificamente attribuibile al deficit di una popolazione di neuroni uditivi o di una via neurale. Questa situazione sta evolvendo rapidamente. I neuroni del sistema nervoso uditivo nel tronco cerebrale sono generati, allo stadio embrionale, da strutture particolari, le labbra rombiche, che generano in particolare delle popolazioni di neuroni destinate al nucleo cocleare. Questo nucleo ha un ruolo essenziale nello smistamento delle informazioni provenienti dal nervo uditivo e nel suddividerle in diverse vie parallele ascendenti. Queste vie trattano specificamente aspetti molto vari del messaggio acustico, quali l’intensità, la struttura temporale fine, i contrasti oppure i rapporti segnale su rumore. L’interessamento specifico di una particolare popolazione è quindi in grado di indurre deficit percettivi, ma le eventuali relazioni anatomocliniche sono ancora speculative. Alcuni modelli animali molto precisi di alcuni deficit centrali iniziano a essere pubblicati. Per esempio, una cattiva regolazione di una via di secrezione di sostanze derivate dall’FGF (fibroblast growth factor) può alterare la struttura dei nuclei uditivi, come nel caso di alcune anomalie di espressione del Sef descritte recentemente; Sef è un antagonista dell’FGF [73]. I topi hanno una dismorfosi del nucleo cocleare e soglie uditive normali, ma PEU anormali. Questo modello può servire a capire meglio alcuni deficit uditivi centrali, ancora poco studiati. Bambini affetti dalla malattia di Gaucher, malattia da sovraccarico associata a una disfunzione dei lisosomi, hanno in effetti un fenotipo uditivo simile a quello descritto nei topi Sef [74, 75].
Un altro modello recente e quello dell’anomalia del fattore di trascrizione Math5 [76] necessario allo sviluppo del nervo ottico, ma che si esprime pure nel sistema uditivo centrale. Nel tronco cerebrale adulto, e in particolare nel nucleo cocleare ventrale, Math5 è espresso da una sottopopolazione che si proietta verso il nucleo mediano del corpo trapezoide, il complesso olivare superiore laterale e il lemnisco laterale. Questa sottopopolazione è in particolare composta da neuroni di tipo globulare e da piccole cellule a cespuglio. I topi mutanti hanno un nucleo cocleare troppo piccolo e i PEU presentano delle latenze aumentate tra i picchi II e IV (omologhi dei picchi III e V nell’uomo). Gli autori, avendo questi neuroni e le strutture alle quali appartengono un ruolo importante nella localizzazione, ritengono senza averne al momento la prova che questa funzione sia probabilmente alterata nei mutanti. D’altra parte, si conosce nell’uomo la sindrome HGPPS (horizontal gaze palsy and progressive scoliosis) nella quale le vie nervose che collegano il nucleo cocleare e il nucleo mediano del corpo trapezoide sono malformate [77] con PEU anormali. Tuttavia, non sembra evidente che i pazienti HGPPS (molto rari) abbiano problemi di localizzazione uditiva; è però vero che questo elemento non è mai stato valutato.
L’importanza di questi studi nell’animale va al di là dell’interesse di etichettare precisamente sindromi molto rare, è anche di comprendere meglio le particolarità dei CAPD (central auditory processing disorders), nei quali i pazienti colpiti hanno difficoltà centrali e difficoltà di discriminazione o di localizzazione [78]. Il concetto di CAPD (descritto oltre) al momento raggruppa indiscriminatamente molte patologie, ma è probabile che sarà suddiviso in elementi di base che corrispondono ad alterazioni specifiche dei circuiti o delle popolazioni neuronali uditive; gli studi genetici permettono di manipolare a volontà queste anomalie e di studiare le loro conseguenze funzionali, modello dopo modello.
|
71] |
Delmaghani S., Del Castillo F.J., Michel V., Leibovici M., Aghaie A., Ron U., e al. Mutations in the gene encoding pejvakine, a newly identified protein of the afferent auditory pathway, cause DFNB59 neuropathyNat. Genet. 2006 ; 38 : 770-778 [cross-ref] |
|
Ebermann I., Walger M., Scholl H.P., Charbel Issa P., Lüke C., Nurnberg G., e al. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction Hum. Mutat. 2007 ; 28 : 571-577 [cross-ref] |
|
|
Abraira V.E., Hvun N., Tucker A.F., Collinq D.E., Brown M.C., Lu C., e al. Changes in Sef levels influence auditory brainstem development and function J. Neurosci. 2007 ; 27 : 4273-4282 [cross-ref] |
|
|
Kaga K., Ono M., Yakumaru K., Owada M., Mizutani T. Brainstem pathology of infantile Gaucher’s disease with only wave I and II of auditory brainstem response J. Laryngol. Otol. 1998 ; 112 : 1069-1073 |
|
|
Lacey D.J., Terplan K. Correlating auditory evoked and brainstem histologic abnormalities in infantile Gaucher’s disease Neurology 1984 ; 34 : 539-541 |
|
|
Saul S.M., Brzezinski J.A., Altschuler R.A., Shore S.E., Rudolph D.D., Kabara L.L., e al. Math5 expression and function in the central auditory system Mol. Cel. Neurosci. 2008 ; 37 : 153-169 [cross-ref] |
|
|
Amoiridis G., Tzagournissakis M., Christodoulou P., Karampekios S., Latsoudis H., Panou T., e al. Patients with horizontal gaze palsy and progressive scoliosis due to ROBO3 E319K mutation have both uncrossed and crossed central nervous system pathways and perform normally on neuropsycholgical testing J. Neurol. Neurosurg. Psychiatry 2006 ; 77 : 1047-1053 [cross-ref] |
|
|
Gopal K.V., Kowalski J. Slope analysis of auditory brainstem responses in children at risk of central auditory processing disorders Scan. Audiol. 1999 ; 28 : 85-90 [cross-ref] |
|
|
Thai-Van H., Micheyl C., Moore B.C., Collet L. Enhanced frequency discrimination near the hearing loss cut-off : a consequence of central auditory plasticity induced by cochlear damage? Brain 2003 ; 126 : 2235-2245 [cross-ref] |
|
|
Willeford J. Differential diagnosis of central auditory dysfunction Audiology : an audio journal for continuing education New-York: Grune and Stratton (1976). |
Ci sono fattori di rischio per la neuropatia uditiva?
I fattori di rischio possono rientrare in tre categorie: a)patologie neonatali, b)processi infiammatori, c)condizioni genetiche o sindromiche. Fra i primi a)sono particolarmente importanti le condizioni di ipossia e di iperbilirubinemia. Questi fattori di rischio includono ittero, parto prematuro, basso peso alla nascita e un inadeguato apporto di ossigeno al feto. Inoltre, alcuni farmaci che sono stati utilizzati per il trattamento complicazioni mediche nelle donne in gravidanza o neonati possono danneggiare le cellule ciliate interne nelle orecchie del bambino, causando neuropatia uditiva. Fra i secondi b)sembrano particolarmente implicati il morbillo e le meningiti. Fra i fattori genetici 3)rientrano alcune forme con manifestazioni generalizzate di neuropatia motoria e sensitiva come la sindrome di Charcot-Marie-Tooth (degenerazione mielinica) e la forma già ricordata da alterazione della otoferlina. Altre forme descritte in associazione ad una AN/AD sono l’atassia famigliare di Friedreich (neuro degenerazione tronco-cerebellare in cui possono essere interessati anche i neuroni del ganglio spirale), sindromi da disordine immunitario (sindrome di Grillane-Barrè) e malattie mitocondriali. Diversi fattori sono stati collegati a neuropatia uditiva nei bambini.
Disturbi associati e diagnosi differenziale
La Neuropatia uditiva è stata segnalata in varie neuropatie ereditarie. Berlin et al (1994) hanno descritto una neuropatia uditiva in tre pazienti affetti da Charcot-Marie-Tooth, che è una sindrome con neuropatia ereditaria. Doyle, Sininger e Starr (1998) hanno riportato un soggetto con Atassia di Fredreich,che è un atassia ereditaria, associata a neuropatia. Bamiou e altri (2003) hanno descritto perdite uditive con ABR anormali nella malattia di Refsum – che è una malattia neurologica molto rara associata a la retinite pigmentosa, polineuropatia, dermatite, anosmia e perdita dell’udito. Papadakis et al. (2003) hanno segnalato un caso di ipoacusia bilaterale attribuita alla malattia di Charcot-Marie-Tooth. Starr e gli altri (1996) hanno inoltre riferito 3 pazienti con neuropatia ereditaria. Neuropatia uditiva può verificarsi anche in forma familiare isolata (Wang et al, 2003).
La Neuropatia uditiva è stata riportata anche nell’ AIDS (HIV). Non tutti i pazienti con AIDS sviluppano perdita dell’udito, ma (Sooy 1987; Bell, Atkins et al. 1988; Kantu, Lee et al. 1996)riportano tassi dal 20,9% al 49%,Questa sindrome è caratterizzata da perdita dell’udito alle alte frequenze. Nell’AIDS la neuropatia uditiva viene di solito diagnosticata da latenze ABR allungate e forme d’onda atipiche (Belman, Ultmann et al. 1985; Geltma e Schüpbach 1986; Smith, Jakobsen et al. 1988)
Si dovrebbe effettuare la diagnosi differenziale con le malattie autoimmuni dell’orecchio interno ,con i pazienti con la sordità improvvisa, effettivamente in questi casi la NU è incerta. Naturalmente, questo potrebbe essere diagnosticata con le ‘ OAE .
La Neuropatia Uditiva può anche influenzare la funzione vestibolare (Sheykholeslami et al. 2000; Fujikawa e Starr, 2001). Quando la neuropatia uditiva è associata alla neuropatia vestibolare, la diagnosi viene fatta combinando i consueti criteri di neuropatia uditiva con le anomalie nei test di funzionalità vestibolare. È probabile che alcune persone con insufficienza vestibolare bilaterale potrebbe avere questo patologia a causa della neuropatia uditiva,e un maggiore uso dei test ABR e dei test VEMP potrebbe essere utile in questa popolazione.
Ci si potrebbe aspettare che il diabete, che è comunemente associato a neuropatia dei nervi cranici potrebbe anche essere associata alla neuropatia uditiva. Questa associazione, finora, non sembra documentata in letteratura. Allo stesso modo, la sindrome di Guillain Barre e la sindrome di Miller Fisher (due tipi di neuropatie acquisite), probabilmente hanno qualche incidenza nella presenza di neuropatia uditiva . Un maggior uso di ABR e di emissioni otoacoustiche (OAE), in questo contesto potrebbe essere utile il test VEMP (una prova di risposta vestibolare evocata).
Come viene diagnosticata la neuropatia uditiva?
Gli operatori sanitari, tra cui otorinolaringoiatri (orecchio, naso, gola) e medici, pediatri, e audiologi, utilizzano una combinazione di metodi per la diagnosi di neuropatia uditiva. Questi prove comprendono le risposte uditive del tronco encefalico (ABR) e le otoemissioni acustiche (OAE).
Diagnosi elettrofisiologica caratterizzato da:
- Presenza di emissioni acustiche Evocati Oto?
- Assente o anormale ABR con microfonicità cocleari presente?
- Riflessi stapediali assenti?
- Risposte comportamentali variabili
Starr et al (1996), Berlino CI, Morlet T, Cappuccio LJ (2003)
Soglia tonale. Nella AN/AD la soglia per toni puri può variare dalla normalità fino a livelli corrispondenti alla sordità profonda. Nel 70% dei casi risulta innalzata oltre 35 dB HL, e metà di questi mostra livelli di soglia oltre 70 dB HL. Sono state frequentemente osservate significative fiuttuazioni uditive, con variazioni temporanee di circa 20 dB. Si è osservato inoltre che in una modesta proporzione di casi la soglia tonale va incontro ad un peggioramento progressivo. Sono stati descritti anche casi in cui la soglia uditiva dimostra un progressivo miglioramento, talvolta fino a livelli di normalità. Ciò sembra accadere con maggiore probabilità in età infantile.
I profili di soglia sono per lo più simmetrici, ed in circa un terzo dei casi sono ascendenti, con un maggior interessamento per le frequenze medio-gravi.
Distorsioni percettive
I profili della percezione uditivo dei pazienti con AN/AD sono diversi da quelli che si osservano nei pazienti con cocleopatia Mentre questi manifestano tipicamente distorsioni di loudness e della risoluzione delle frequenze, nei casi con AN/AD
Riflessi stapediali. I riflessi stapediali per stimolazione ipsi- controlaterale sono assenti. indipendentemente dal livello della soglia per toni puri. L’assenza dei riflessi stapediali è spiegata con un insufficiente sincronismo di scarica dei neuroni afferenti che proiettano sul nucleo motore del n. VII. Infatti i riflessi non-acustici (da stimolazione tattile della faccia) rimangono conservati, indicando che la porzione efferente dell’arco riflesso è normale.

Fig. 3: Tympanometry- bilaterale di tipo “A” di curva, senza ipsi e controlaterale riflesso acustico. (Impression: Nessuna indicazione di patologia dell’orecchio medio)
Otoemissioni acustiche (OAE)
OAE è generato attivamente dalla motilità delle cellule ciliate esterne in risposta ad uno stimolo in entrata.
Importanza OAE
• Richiede la normale funzione dell’orecchio esterno, medio ed interno – fino alle cellule ciliate esterne.
• Non registrabile quando soglia media> 20-30dB
• Indica soddisfacente funzione uditiva periferica.
OAE. Le OAE sono normali nella AN/AD. Si ricorda che le OAE sono l’espressione di un processo affivo delle cellule cigliate esterne, ritenuto essenziale per garantire un’elevata sensibilità uditiva ed una fine sintonia fra forme acustiche e forme meccaniche della membrana basilare. Normalmente, se la soglia uditiva è inferiore a 20 dB HL le OAE sono presenti al 99%. Nelle sordità neurosensoriali le OAE da transitori (TEOAE) sono assenti con perdite uditive oltre i 35 dB HL, mentre le OAE come prodotti di distorsione (DPOAE) sono assenti con perdite uditive oltre 60 dB HL. La presenza di queste risposte è pertanto indicativa di una normalità funzionale dell’amplificatore cocleare.
MC. Il ruolo del microfonico cocleare (MC) nella diagnosi di NA/ND è controverso Si ritiene che il MC, potenziale di recettore e quindi preneurale, origini da fenomeni elettrici di membrana delle cellule cigliafe. Per alcuni la presenza di MC associata all’assenza di ABR sarebbe una conferma di NA/ND Per altri il valore diagnostica del MC sarebbe scarso, dal momento che esso risulta sempre registrabile. anche nei casi di ipoacusia profonda.

Figura 4 (a, b): Emissione Otoacostiche (OAE) – Le TEAOE e DPOAE- mostrano una funzione cocleare normale bilaterale
Elettrococleografia (EcochG) & elettrico ABR (EABR) nella diagnosi di neuropatia uditiva (case study)

ABR. L’assenza di ABR alla massima intensità di stimolazione (90 dB nHL) anche nei casi con una soglia tonale relativamente conservata è attualmente giustificata con due ipotesi a) una diminuzione di neuroni disponibili all’attivazione con una conseguente riduzione d’ampiezza dei potenziali evocati, così da non poter essere risolti dai comuni dispositivi di registrazione (rapporto segnale/rumore eccessivamente piccolo);
 b) una disorganizzazione temporale dell’attivazione neurale, così che i singoli potenziali di fibra, non più sincroni, non riescono a “costruire” il potenziale uditivo registrabile in superficie.
b) una disorganizzazione temporale dell’attivazione neurale, così che i singoli potenziali di fibra, non più sincroni, non riescono a “costruire” il potenziale uditivo registrabile in superficie.
Figura 4: ABR- uditiva del cervello response- non definita onda V sono stati visti su entrambi i lati, anche dopo 90 dB e 96 dB.
Ulteriori risultati
|
Soglie di PTA |
Normale per sordità profonda, può essere asimmetrica, una configurazione variabile |
|
Il riconoscimento vocale in posizione tranquilla |
Variabile: da lievi a gravi problemi |
|
Parlato nel rumore |
povero |
|
OAE |
Normale inizialmente, ma può scomparire in seguito |
|
Soppressione OAE |
assente |
|
Microfonici cocleari |
Present (inverte quando stimolo polarità è invertita – Berlino et al 1998) |
|
Riflessi stapediale |
assente |
Patologia
Disfunzione delle cellule ciliate interne Fig.5.
Disfunzione della sinapsi Fig.6.
VIII nervo – vero neuropatia uditiva Fig.7.
Anomalie del tronco encefalico Fig.8.
AN / AD comprende una serie di condizioni che si differenziano per sito e patologia – Cone-Wesson e Rance, 2000, Starr et al, 2000).
|
|
|
|
|
Fig.5. cellule ciliate interne assenti o malfunzionanti
|
Fig.6. Disfunzione della sinapsi tra l’IHC e VIII N (problema trasmettitore)
|
Fig.7. VIII nervo – VerA neuropatia uditiva *
|
|
|
|
|
|
Fig.8* ” Neuropatia Uditiva ” è un processo patologico che coinvolge il nervo cranico VIII Vera neuropatia che coinvolge il nervo VIII
|
Fig. 9 |
|


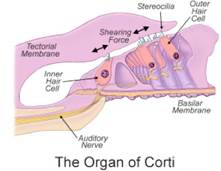


Fig.10 Le forme d’onda I primi possono essere normale
|
Hearing Loss categoria |
ECochG |
acABR |
EABR |
|
Perdita sensoria |
Da lieve a soglie prof |
Presente / normale per lievi a perdite moderatamente gravi |
Presente / normale per gravi profonde perdite |
|
IHC / Synapse |
APP a una o più frequenze |
Assente / forma d’onda anomala |
Normale |
|
Vero uditiva Neuropatia |
APP a una o più frequenze |
Assente / forma d’onda anomala |
Ritardato onda V |
|
Uditivi del tronco encefalico Neuropatia |
APP a una o più frequenze |
Assente / forma d’onda anomala |
Onde Primi presente ma onda V in ritardo / assente |
Caratteristiche associate
- Modello di perdita dell’udito neurosensoriale – spesso a bassa frequenza (Sininger)
- Fluttuante soglie uditive
- Riflessi acustici Assente
- Perdita di discriminazione, soprattutto alle basse frequenze
- Diffusione anomala di mascheramento
- Particolari difficoltà con discorso-in-rumore
- Elaborazione temporale Poor / rilevazione Poor divario
- Localizzazione Poor – ridotta capacità di rilevare interaurale differenze temporanee
- Risposte neurali Dys-sincroni

Da: conseguenze percettive di attività nervo uditivo perturbato, Zeng FG, Kong YY, Michalewski HJ, Starr A, J Neurofisiologia 93: 2005
AD come parte della neuropatia generalizzata
Riportato in
- Ereditarie Motor & neuropatie sensoriali [cioè di Charcot Marie Tooth, CMT]
- Di Friedreich [FA]
- X eredità legata – AUNX1
- Diabete mellito
CMT-Mutazione di PMP22 gene che è essenziale per la formazione della mielina – posizione cellule di Schwann – ereditarietà dominante.
neuropatia uditiva Unilaterale…
- Ngo, 2006: studio di AD / AN in un programma di screening uditivo neonatale, Singapore.
- 9/52 bambini affetti da ipoacusia avevano AD / AN [6 bilaterale e unilaterale 3]
- Quelle unilaterali non aveva perinatali ‘fattori di rischio’, una aveva Moebius, uno aveva assente ottavo nervo
- Isolati case report: rilevamento incidentale sullo screening della scuola, uno di seguito parotite.
Ritardato la maturazione visiva e uditiva in AN / AD
Aldosari et al (2003)
- ABR nessuna risposta alle 90dBnHL scatta al momento della nascita
- OAE normali
- Nessun fissazione visiva
- A 9 mesi ABR – normale
Condizioni mediche che possono causare AN / AD
Cause perinatali – Madden et al (2002) 22 pazienti studiati
Iperbilirubinemia 50%
Prematurità 45%
Farmaci ototossici 41%
(Storia di famiglia 36%)
Neonatale Ventilatore dipendenze 36%
Paralisi cerebrale 8%
Quali trattamenti, dispositivi e altri approcci possono aiutare le persone con neuropatia uditiva per comunicare?
Protesizzazione acustica
Quando un AN/AD si manifesta con un innalzamento di soglia si pone il problema di aumentare l’udibilità dei segnali per mezzo di una protesi acustica. Questo indirizzo tuttavia è ancora controverso perché vi sono almeno due aspetti che potrebbero controindicare l’amplificazione. Il primo riguarda il rischio di danneggiare le cellule cigliate esterne della coclea, che nella AN/AD sono normo funzionanti con cromeccanica cocleare conservata che potrebbe risentire di una sovra-applicazione. Il secondo riguarda la modalità con cui vengono generati e trasferiti i pattern neurali, destinata a permanere imprecisa (dis-sincronia) anche se i segnali amplificati sono percepiti come più intensi In tal caso l’amplificazione del parlato non migliorerebbe l’intelligibilità. Per verificare questa evenienza si raccomanda di testare l’intera funzione di intelligibilità. una funzione che tende ad appiattirsi suggerisce che l’amplificazione avrà uno scarso beneficio, mentre una funzione con roll-over suggerisce che l’amplificazione sarà controproducente
Nei bambini con AN/AD l’amplificazione diventa in ogni caso necessaria per garantire I’udibilità dell’informazione associata al parlato In questi casi si raccomandano valori di guadagno più ridotti rispetto a quanto previsto dalle formule prescrittive. In alcuni pazienti la protesizzazione potrebbe essere solo monolaterale, per semplificare il processamento centrale.
In generale, la protesizzazione nell’adulto con AN/AD, anche se accettata, si è dimostrata deludente quando si testa l’intelligibilità verbale. Nei bambini i risultati sono più variabili perché in certi casi è stato dimostrato un vantaggio significativo nell’ascolto con protesi, tuttavia i livelli della prestazioni tendono sempre a rimanere al di sotto di quelle dei bambini con sordità neurosensoriale da cocleopatia. Alcune casistiche stimano che circa la metà dei bambini con AN/AD non hanno alcun vantaggio dall’amplificazione.
Impianti cocleari
Molti casi di AN/AD sono stati trattati con impianto cocleare, con benefici spesso confrontabili ai tipici casi di sordità profonda. Dati i limitati vantaggi dell’amplificazione oggi si pone il problema di un’indicazione all’impianto anche nei casi di AN/AD con sordità moderata-severe e generalmente senza particolari problemi durante l’attivazione. Anche l’evoluzione delle abilità percettive si sviluppa, nei bambini, in modo simile ai casi di sordità neurosensoriale. Il miglioramento delle prestazioni di ascolto ottenute con l’impianto cocleare è legato da un lato alla percezione, mediata dalla stimolazione elettrica. di un ampio spettro di stimoli acustici. ma anche dal fatto che probabilmente la stimolazione elettrica facilita la costituzione di pattern neurali più precisi, contrastando in qualche misura la condizione di dis-sincronia lnfatti la modalità di stimolazione impulsiva fornita al nervo potrebbe di per sé produrre pattern neurali più sincroni Un’altra spiegazione, valida per i casi di AN/AD sostenuti da una lesione selettiva delle cellule cigliate interne, può essere che la stimolazione elettrica aumenti il numero di fibre che si attivano, by-passando il blocco cito-neurale. Un test ritenuto predittivo nei confronti dell’efficacia dell’impianto cocleare è la registrazione dell’ABR a stimoli elettrici La presenza di un ABR normale indica che la via uditiva è in grado di rispondere con una normale sincronia di scarica quando attivata da impulsi elettrici
La fig. 13-14 riporta il caso di un bambino con AN/AD. L’elettococleografia effettuata a 17 mm di età mostra un ampio MC, di durata prolungata, ed un potenziale d’azione di morfologia alterata, per la sovrapposizione di quote di potenziale di sommazione o per una dis-sincronia della risposta neurale. L’ABR alla massima intensità di stimolazione è assente. La protesizzazione bilaterale ha ripristinato l’udibilità. ma ciò non ha prodotto risultati nello sviluppo del linguaggio. A 30 mesi di età il bambino ha ricevuto un impianto cocleare nell’orecchio sinistro. Il controllo dopo 18 mesi dall’IC, oltre ad evidenziare l’acquisizione di linguaggio, ha mostrato un significativo miglioramento della soglia tonale a destra.
Impianti cocleari e ANSD
- Panoramica Ricerca
- Risultati
- Gestione
1. Panoramica di prove
- AN bambini fanno beneficiare di CI (scialuppa et al 2001)
- UN v SNHL bambini con CI (Peterson et al 2003)
- AN v AD, Gardner-Berry Gibson & Sanli 2005
- CI HA v, Rance et al 2008 Pronostico risultati CI, Walton, 2008
Peterson Sommario
- Nessuna differenza in requisiti del programma
- Beneficio funzionale (MAIS) lo stesso
- Entrambi i gruppi in gran parte a programmi di istruzione orale
- Consiglia l’impianto cocleare come una valida opzione … Ma … CARE e un’attenta valutazione.
Buchman et al, 2006
- MRI dovrebbe essere effettuato per AN figli:
- 9 (18%) dei 51 bambini avevano carenza nervo cocleare
- I bambini con AN e nervi sottili possono beneficiare di impianto cocleare
- Argomento di studio
Walton et al, 2008
Fattori predittivi per CI + AN
- Comorbilità mediche
- Abilità cognitive
- Carenza di nervo cocleare
- Anomalie coclea
Rance et al 2008
- CI offre la possibilità di significativo percezione open-set per i bambini che mostrano capacità discriminazione solo limitato a ingresso acustico.
- Alcuni bambini AN / AD possono fare bene con l’amplificazione convenzionale.
- Come possiamo differenziare questi bambini?
Note: Questi autori sollevano alcune questioni importanti in materia di gestione di bambini con AN / AD-tipo perdita di udito. Alcuni (in particolare giovani) i pazienti AN / AD che possono fare bene con l’amplificazione convenzionale, quindi, non occorre prendere in considerazione tutti i bambini con questa forma di perdita dell’udito come un candidato adatto per impianto cocleare Determinazione del livello al quale un / AD soggetto AN potrebbe ragionevolmente aspettarsi un meglio esito percettivo con un impianto cocleare richiede maggiore esperienza e numeri oggetto. I risultati di questo studio suggeriscono che questo livello può essere diverso per i soggetti con AN / AD di quello utilizzato per i candidati SN.
Gardner-Berry, Gibson e Sanli 2005
- Ipoacusia sensoriale; buoni risultati con CI
- Auditory Dys sincronia (AD); buoni risultati previsti con CI
- Auditory neuropatia (AN); risultati meno ottimali previsti con CI
- Uditivi del tronco encefalico neuropatia (BAN); risultati meno ottimali previsti con CI.
Note: SNL AD crede di essere una patologia prevalentemente cocleare; buoni risultati previsti con CI AN coerente con la patologia del nervo uditivo. Risultati meno ottimali previsti con CI BAN Coerentemente con uditivi del tronco encefalico patologia. Risultati meno ottimali previsti con CI.
RIFERIMENTI
· Bamiou DE, Spraggs PR, Gibberd FB, Sidey MC, Luxon LM. Hearing loss in adult Refsum’s disease.Clin Otolaryngol 2003 Jun;28(3):227-30
· Bell, A. F., J. S. Atkins, et al. (1988). Sensorineural hearing loss in AIDS. Fourth International Conference on Acquired Immunodeficiency (AIDS), Stockholm.
· Belman, A. L., M. H. Ultmann, et al. (1985). “Neurological complications in infants and children with acquired immune deficiency syndrome.” Annals of Neurology 18(5): 560-6.
· Berlin CI, Hood LJ, Hurley A, Wen H. Contralateral suppression of otoacoustic emissions: an index of the function of the medial oliovocochlear system. Otolaryngol HNS 1994:110:3-21
· Doyle KJ, Sininger Y, Starr A. Auditory neuropathy in childhood. The laryngoscope, 108:1374-1377, 1998
· Fujikawa S, Starr A. Vestibular neuropathy accompanying auditory and peripheral neuropathies. Arch Otolaryngol HNS 2000:126:1463-1456
· Geltma, C. L. and J. E. Schupbach (1986). “Neuro-audiologic findings of a patient with acquired immune deficiency syndrome.” ASHA 26: 76.
· Gibson, W. P. and H. Sanli (2007). “Auditory neuropathy: an update.” Ear Hear28(2 Suppl): 102S-106S.
· Kantu, S., D. Lee, et al. (1996). “Safety awareness for the otolaryngologist caring for the HIV-positive patient.” Laryngoscope 106(8): 982-6.
· Kraus N, Ozdamar O, Stein L, Reed N. Absent auditory brain stem response: peripheral hearing loss or brain stem dysfunction. Laryngoscope 1984:94:400-6
· Liang F et al. Auditory neuropathy: clinical study of 17 cases. ARO abstracts, 2001, #53
· McMahon CM and others. Frequency-Specific Electrocochleography Indicates that Presynaptic and Postsynaptic Mechanisms of Auditory Neuropathy Exist. Ear and Hearing 2008:29(3) 314-325
· Madden C and others. Clinical and audiological features in auditory neuropathy. Arch Otolaryngol HNS 2002:1026-30
· Papadakis CE, Hajiioannou JK, Kyrmizakis DE, Bizakis JG . Bilateral sudden sensorineural hearing loss caused by Charcot-Marie-Tooth disease. J Laryngol Otol 2003 May;117(5):399-401
· Sheykholeslami K, Kaga K, Kaga MJ Laryngol Otol 2001 Jul;115(7):530-4 An isolated and sporadic auditory neuropathy (auditory nerve disease): report of five patients.
· Sheykholeslami K, Kaga K, Murofushi T and Hughes DW (2000). “Vestibular function in auditory neuropathy.” Acta Otolaryngol 120(7): 849-54.
· Smith, T., J. Jakobsen, et al. (1988). “Clinical and electrophysiological studies of human immunodeficiency virus-seropositive men without AIDS.” Annals of Neurology 23(3): 295-7.
· Sooy, C. D. (1987). Impact of AIDS on otolaryngology head and neck surgery. Chicago, Year Book Medical Publishers.
· Starr A. and others. Transient deafness due to temperature sensitive auditory neuropathy. Ear and Hearing, 1998:19:167-179
· Starr A and others. Auditory Neuropathy. Brain (1996) 119, 741-753
· Stein LK and others. Auditory neuropathy associated with elevated bilirubin levels. ARO, 1997
· Stein LK and others. Brainstem abnormalities in neonates with normal otoacoustic emissions. Seminars in Hearing, 17, 2, 1996, 197-213
· Wang DY, Bu XK, Xing GQ and Lu L (2003). “[Neurophysiological characteristics of infants and young children with auditory neuropathy].” Zhonghua Yi Xue Za Zhi 83(4): 281-4.
· Wang Q, Gu R, Han D, Yang W. Familial auditory neuropathy. Laryngoscope. 2003 Sep;113(9):1623-9.
APPROFONDIMENTO
NEUROPATIA UDITIVA ED ELETTROCOCLEOGRAFIA
R. Santarelli E Arslan Percezione Uditiva e Patologie del Linguaggio Omega Edizioni 2013
La neuropatia uditiva (NU) è caratterizzata da ipoacusia bilaterale di entità variabile (da lieve a profonda), che si accompagna a impedenzometria normale e assenza di riflesso stapediale sia ipsi che controlaterale, assenza o presenza di anomalie rilevanti del tracciato ABR e otoemissioni presenti Inoltre generalmente i pazienti presentano una discriminazione verbale peggiore in relazione alla soglia audiometrica.
E’ un disordine dovuto ad una lesione del sistema uditivo periferico, con integrità del sistema delle cellule ciliate esterne. Non c’è accordo su quale sia l’esatta sede di lesione, che potrebbe essere a livello o delle cellule ciliate interne, o delle sinapsi tra le cellule ciliate interne e i dendridi delle cellule gangliari, o delle fibre dell’VIII n. cranico; per questo motivo, è logico ritenere che la NU non sia in realtà un unico quadro patologico, ma un insieme eterogeneo di disordini, in cui riveste un ruolo importante anche una desincronizzazione della risposta del sistema uditivo periferico.
La sua incidenza è maggiore nei neonati ricoverati presso i Centri di terapia intensiva neonatale. Alcuni casi sono ad eziologia genetica ed e stato recentemente identificato un gene, il gene OTOF, responsabile di alcuni casi familiari (DFNB 9); inoltre la NU è stata descritta in pazienti, sia adulti che in età pediatrica, con malattie mitocondriali.
I pazienti affetti da NU ed in particolare i bambini presentano problematiche audioprotesiche di difficile soluzione; infatti la risposta alla protesizzazione acustica tradizionale è variabile ed in molti casi modesta, per quanto riguarda la percezione verbale. Buoni risultati sono invece stati descritti in numerosi casi di bambini affetti da NU sottoposti ad impianto cocleare.
La Definizione di Neuropatia Uditiva (Auditory Neuropathy, AN)
La prima definizione di neuropatia uditiva si deve ad Arnold Starr e collaboratori che in uno studio pubblicato nel 1996 (Starr et al., 1996) hanno riportato le caratteristiche cliniche di un gruppo di pazienti che presentava una severa compromissione della percezione verbale a fronte di una perdita uditiva di grado lieve o moderato. Tali reperti erano associati alla presenza delle otoemissioni acustiche (otoacoustic emissions, OAEs) e all’assenza delle risposte evocate uditive del tronco encefalico (auditory brainstem responses, ABRs). L’assenza di coinvolgimento del sistema nervoso centrale e il riscontro di una neuropatia periferica nella maggior parte dei pazienti aveva indotto gli autori a ipotizzare che la lesione responsabile dell’alterata percezione verbale e dell’assenza della risposta ABR fosse riconducibile a fenomeni di demielinizzazione delle fibre del nervo uditivo o, in alternativa, a lesioni delle cellule ciliate interne (inner hair cells,11-1Cs) o delle sinapsi interposte, Sulla base di tale ipotesi le basi neurofisiologiche della riduzione della percezione verbale erano da ricercare in una alterata codifica dell’informazione uditiva da parte del compartimento afferente periferico associata alla conservazione della funzionalità delle cellule ciliate esterne (outer hair cells, OHCs).
Dall’epoca della pubblicazione di questo primo studio la definizione di neuropatia uditiva è andata incontro a diverse revisioni. Nella sua accezione più recente essa viene definita come un disordine uditivo caratterizzato da una alterazione della codifica temporale dell’informazione acustica a livello delle fibre del nervo uditivo con conseguente compromissione delle funzioni percettive basate sull’analisi temporale (Zeng et al, 2005; Starr et al, 2008). I meccanismi alla base delle modi‑
fiche della scarica neurale comprendono lesioni di tipo pre-sinaptico e post-sinaptico, che causano alterazioni della generazione del potenziale di recettore nelle IHCs, compromissione del rilascio del glutamato dalle sinapsi a nastro, difetto di
innesco del potenziale di azione in corrispondenza della porzione terminale degli assoni e riduzione della velocità di conduzione lungo le fibre nervose. Tali meccanismi, agendo separatamente o in associazione, determinano invariabilmente la riduzione del sincronismo di attivazione delle fibre neurali (Fig. 1A).
|
|
|
Fig. 1A i possibili meccanismi aal base delle forme pre-sinaptiche e post-sinaptiche di neuropatia uditiva, Nei riquadri sono elencati i geni implicati nei disordini genetici associati a ciascuna forma. |

Epidemiologia
La prevalenza della neuropatia uditiva varia tra 1’1% e il 10% (Berlin et al, 2010). La notevole variabilità delle stime riportate riflette verosimilmente l’inclusione di malattie (per es. sindrome di Guillain-Barré) in cui il disordine uditivo può essere solo transitorio (Kaga e Starr, 2009). Inoltre, nelle stime di incidenza vengono generalmente compresi i disordini che interessano i neonati ricoverati nelle unità di terapia intensiva neonatale (TIN). In questi piccoli pazienti il segno distintivo dell’AN é rappresentato dall’assenza della risposta ABI associata al riscontro delle otoemissioni acustiche. ln questi casi, tuttavia, le alterazioni dei potenziali del tronco potrebbero risultare da un ritardo di maturazione del nervo uditivo e dei generatori tronco-encefalici (Kraus et al, 1984), configurando pertanto delle condizioni transitorie o comunque non inquadrabili in un quadro di neuropatia uditiva vero e proprio. D’altra parte, altre forme di neuropatia uditiva potrebbero non essere state incluse nelle stime di prevalenza per le difficoltà diagnostiche poste talora da alcuni pazienti per i quali le difficoltà della percezione verbale si evidenziano unicamente in presenza di un segnale competitivo (Starr et al, 2008).
La neuropatia uditiva può essere congenita o acquisita. Le forme congenite interferiscono con lo sviluppo del linguaggio, il quale dipende criticamente dalla presenza di un input uditivo efficiente che guidi l’organizzazione del sistema uditivo durante la fase di massima plasticità cerebrale (Johnson e Newport, 1991). L’insorgenza del disordine in epoca post-verbale, in età scolare o al più tardi durante l’adolescenza, determina la compromissione della percezione verbale associata all’eventuale regressione delle abilità linguistiche nel loro complesso.
Sia le forme congenite che quelle acquisite riconoscono una causa genetica
o possono essere dovute a fattori eziologici di vario tipo quali malattie del sistema immunitario, cause tossiche e malattie metaboliche(Starr et al, 2000; Santarelli et al, 2006; Santarelli et al, 2008; Starr et al, 2008). Tuttavia, in almeno la metà dei casi l’eziologia rimane sconosciuta (Starr et al, 2008).
Le varie forme di questo disordine si possono presentare isolatamente (isolated AN) o in associazione al coinvolgimento di altri organi o apparati (non isolated AN), Sicuramente le cause di tipo genetico rappresentano ad oggi i fattori eziologici più frequentemente associati alle varie forme di AN, I disordini genetici ad oggi noti, implicati nella eziopatogenesi sono elencati nella Tab. 1 e sono stati distinti in forme isolate e non isolate. Per ciascuna forma sono riportate le caratteristiche fenotipi-che più frequenti, inclusi il grado di perdita uditiva più frequentemente riscontrato e il tipo di patologia eventualmente associato. Per quel che riguarda le forme non isolate, il riscontro predominante è rappresentato dalla neuropatia ottica e dalle neuropatie periferiche, che possono presentarsi a loro volta come forme demielinizzanti, assonali e miste (Santarelli, 2010).
Caratteristiche cliniche e fisiopatologia
I criteri clinici per una prima formulazione diagnostica consistono essenzialmente nel grave ritardo dello sviluppo linguistico nelle forme congenite, e nella severa compromissione della percezione verbale nelle forme acquisite, condizioni entrambe associate alla destrutturazione della risposta ABI e al riscontro delle otoemissioni acustiche.
Auditori, Neurapathy associated with Genetic Diseases
|
|
Locus |
Gene |
Transmission |
Phenotype |
Reference |
|
isoiated AN |
|
|
|
|
|
|
|
2p23-p22 |
OTOF |
Recessive |
Congenital profound deafness |
Varga, 2003 |
|
|
431.1-01,3 |
PAIK |
Recessive |
Congenitat profound deafness |
Dei maghe ni, 2006 |
|
|
13q21-q24 |
WAPH3 |
Dominant |
Moderate to profound deafness |
Les pe ra ne, 2010 |
|
|
mtONA |
125 rRNA |
|
Moderate deafness |
Wang, 2005 |
|
Non-isoiated AN |
|
|
|
|
|
|
CMT lA |
17p11.2-p12 |
PMP22 |
Dominant |
M itcl to severe deafness; demyelEnaUng neuropathy |
Kovach, 2002 |
|
CMT 1B |
1822 |
MPZ |
Dominant |
Mild to severe deafness; demyetfnating neuropathy |
Sun’, 2003 |
|
CMT 2E |
8;321 |
NF.4.. |
Dominant |
Normal hearing; akonal neuropathy |
gutlner, 2003 |
|
CMT 40 |
3o24.3 |
NORG2 |
ReceSsive |
M ild to severe deafness; axonal/demyelinating neuropathy |
Kai aydileva, 2000 |
|
CMT |
1p34 |
6/33 (Cx31) |
Dominant |
M il d deafness |
1,4pez-81gas, 2001 |
|
CMT 1X |
N}13 |
G/31 (Cx32) |
X-linked |
Demyetinating neuropathy |
Rahr, 1099 |
|
|
|
|
Dominant |
|
|
|
A DOA |
3o28-O29 |
PPAZ (R4451-) |
Dominant |
Qptic neuropathy; moderate deafness |
Amati‑ |
|
|
|
|
|
|
8onnead,2005 |
|
ARDA |
11814.1— |
TMEM226A |
Recessive |
Optic neuropathy; mltd hearíng iosa |
Meyer, 2010 |
|
|
11q22,3 |
|
|
|
|
|
Friedre I ch |
903 |
FXN |
Recessive |
Atarb; axonal neuropathy; opttc neuropathy; ordiornyopathy; normal licadng threshdtd-mil/ deafness |
Rance et al, 2008 |
|
AUNX1 |
xq23-q27.3 |
|
Minked |
Sensory axonal neuropallty; rnitd.to- severe deafness |
Wang et al. 2005 |
|
DDON (Mo h r -Tr a n e 1›.) LHON (Le ber) |
X822.1 mtDNA |
17MM9A |
X-II nked Recessive |
ProgreSstve deafrtess; dystonia, op ti c neuropathy; dern colla Ootic neuropathy; rnild-to-moderate |
Bah rn ad, 2007 |
Un tipico esempio è rappresentato dalle forme che si ritrovano in associazione alle neuropatie periferiche demielinizzanti del gruppo Charcot-Marie-Tooth, quali quelle sottese da mutazioni puntiformi dei geni che codificano per due distinte proteine associate alla mielina, la proteina MPZ (Starr et al, 2003) e la PMP22 (Kovach et al, 2002). Esami autoptici effettuati in pazienti con mutazione nel gene che codifica per MPZ hanno rivelato estese lesioni a carattere demielinizzante e perdita neuronale a carico del nervo uditivo analoghe a quelli osservate in corrispondenza dei nervi somatici (neuropatie assonali e demielinizzanti). Al contrario, le cellule ciliate cocleari risultavano indenni. In questi casi, la destrutturazione dei potenziali del tronco è solo un effetto del problema elettrofisiologico di base, che risulta dall’alterazione della velocità di conduzione nelle fibre nervose. Ne risulta una riduzione globale dell’input uditivo e del suo livello di sincronizzazione con conseguente impossibilità da parte del sistema nervoso centrale di utilizzare la modalità di codifica temporale ai fini della percezione uditiva e verbale. Il corrispettivo psicoacustico è rappresentato da un innalzamento preferenziale della soglia uditiva alle frequenze gravi associato a una marcata riduzione della capacità di individuazione delle rapide fluttuazioni di intensità e frequenza del parlato con conseguente gravissima compromissione della percezione verbale,
Analoghe alterazioni psicoacustiche ed elettrofisiologiche possono derivare da un difetto di innesco del potenziale di azione in corrispondenza della porzione terminale degli assoni. Alterazioni della genesi del potenziale di azione per lesioni che coinvolgono la porzione dendritica delle fibre uditive costituiscono il meccanismo patogenetico ipotizzato nel caso dell’atrofia ottica dominante (DOA) dovuta nella maggior parte dei casi a mutazioni puntiformi localizzate nel gene OPA1 (Huang et al, 2009). OPA1 è una proteina mitocondriale codificata a livello nucleare e localizzata a livello della superficie interna dei mitocondri, che risulta coinvolta in vari processi quali la fosforilazione ossidativa, i processi dl fusione e il mantenimento dell’integrità delle creste mitocondriali (Delettre et al, 2000; Olichon et al, 2003; Frezza et al, 2006). Il disordine insorge in età infantile con una riduzione della capacità visiva dovuta alla progressiva atrofia del nervo ottico seguita dall’instaurarsi del disordine uditivo anch’esso con caratteri di progressione (Kjer, 1959). Si ritiene che il deficit uditivo e visivo condividano la stessa patogenesi e riguardino cioè primariamente le terminazioni amieliniche del nervo ottico e del nervo uditivo caratterizzate da una elevata richiesta energetica dovuta alla conduzione elettrotonica dell’impulso nervoso (Carelli et al, 2004). La forme di AN dovute a lesioni a carattere degenerativo che coinvolgono gli assoni del nervo e la componente dendritica delle fibre uditive costituiscono evidentemente delle patologie post-sinaptiche.
La più tipica delle forme pre-sinaptiche è rappresentata dalla neuropatia uditiva risultante da mutazioni localizzate nel gene che codifica per otoferlina (OTOF, DFNB9)(Rodriguez-Ballesteros et al, 2003; Varga et al, 2003). Questa costituisce una proteina integrale di membrana localizzata al polo sinaptico delle ICHs, che contiene diversi siti ripetitivi leganti il calcio (Rizo e Sűdhof, 1998). Otoferlina svolge un ruolo cruciale nella liberazione delle vescicole a livello delle sinapsi a nastro interagendo con le proteine sintaxinal e SNAP25 (Roux et al, 2006). lnfatti, è stato dimostrato in topi knock-out che la fase rapida iniziale dell’esocitosi è abolita in , assenza della proteina, mentre la fase tardiva risulta sostanzialmente conservata. Inoltre, del tutto recentemente è stato riconosciuto il ruolo dell’otoferlina nel reintegro continuo del pool delle vescicole al polo sinaptico delle IHCs (Pangrsic et al, 2010). Sulla base di questi dati è evidente che le alterazioni della funzione uditiva sottese da una riduzione della funzione di otoferlina rappresentano più propriamente una patologia sinaptica che coinvolge il funzionamento delle sinapsi a nastro. La riduzione della liberazione del neuromediatore associato alla compro-missione del meccanismo cooperativo del rilascio multivescicolare determina una riduzione della sicurezza sinaptica (Singer et al, 2009) con conseguente aumento della probabilità del numero di “rilasci a vuoto”, non seguiti cioè dalla insorgenza di potenziali azione,
Ad oggi sono state identificate più di 40 mutazioni patogene, la maggior parte delle quali sono “truncating mutations”, causano cioè una inattivazione della funzione della proteina (Rodrfguez-Ballesteros et al, 2008), La trasmissione avviene in forma recessiva nella quasi totalità dei casi ed è associata ad un fenotipo sostanzialmente omogeneo caratterizzato da ipoacusia neurosensoriale di grado severo-profondo ad insorgenza preverbale. Almeno la metà dei soggetti affetti presenta le otoemissioni acustiche, ma è verosimile che questo dato rappresenti una sottostima. E’ importante sottolineare che il riscontro delle OAEs in soggetti affetti da ipoacusia di grado elevato rappresenta un problema per tutti i programmi di screening uditivo che utilizzano la registrazione delle otoemissioni acustiche come unico procedimento di identificazione dei soggetti affetti da ipoacusia.
Le mutazioni che non portano a una completa inattivazione dell’otoferlina ma
alla sintesi di una proteina instabile o di ridotta attività (missense mutations) danno luogo a un fenotipo caratterizzato dalle classiche manifestazioni cliniche della
neuropatia uditiva che si presentano in forma evidente in caso di aumento della temperatura corporea (febbre, sforzo fisico eccessivo)(Varga et al, 2006; Wang et al, 2010; Wynne et ai, 2013). E’ verosimile che in questi casi la proteina vada incontro a una instabilità funzionale a temperature corporee più elevate della norma.
Infine, nell’ambito delle neuropatie uditive il ‘gruppo dei neonati dimessi dalla Terapia Intensiva Neonatale (TIN) che presentano le OAEs a fronte di una risposta ABR profondamente alterata, costituisce un problema relativo non solo alla definizione della fisiopatologia e alla individuazione del sito di lesione, ma che attiene più radicalmente alla nozione stessa di neuropatia uditiva che, almeno in un certo numero di casi, potrebbe essere messa ragionevolmente in discussione.
I bambini dimessi dalla TIN presentano un aumentato rischio di ipoacusia neurosensoriale per l’esposizione a tutta una serie di fattori potenzialmente lesivi per la periferia uditiva, quali la prematurità, il basso peso alla nascita, l’ipossia da cause respiratorie, l’anemia, l’iperbilirubinemia, il rumore e i farmaci ototossici (American Academy of Pediatrics, Joint Committee on lnfant Hearing, 2007; Hille et al, 2007). L’incidenza della perdita uditiva in questo gruppo di pazienti varia tra 1.5% (Xoinis et al, 2007) e 3.2% (Hille et al, 2007) nelle diverse casistiche, Le differenze rilevate tra i vari studi sono correlate con le caratteristiche cliniche dei pazienti ricoverati nei vari ospedali, l’età alla diagnosi e la variabilità nelle modalità di accertamento e di definizione del deficit uditivo. Ciò che è veramente importante tuttavia dal punto di vista della fisiopatologia della lesione periferica è tenere conto del fatto che l’effetto combinato dei vari fattori di rischio agisce sulla funzione cocleare in maniera differenziata sulle sue diverse componenti. Ne risultano gradi variabili di perdita uditiva correlati con possibili diversi livelli di coinvolgimento di OHCs, IHCs e fibre del nervo uditivo.
E’ stato stimato che circa il 5.6% dei bambini ricoverati in TIN che risultano “fai]” allo screening uditivo, presentano il tipico quadro “elettrofisiologico” della neuropatia uditiva caratterizzato dall’associazione del riscontro delle CAEs in assenza di risposta ABR (American Academy of Pediatrics, Joint Committee on lnfant Hearing, 2007; Xoinis et al, 2007). Lo studio istopatologico delle coclee prelevate da bambini ricoverati e successivamente deceduti in TIN che dimostravano il tipico profilo elettrofisiologico della neuropatia, ha dimostrato la perdita selettiva delle IHCs con conservazione delle OHCs (Amatuzzi et al, 2011). Sulla base di questi dati è stato suggerito che il quadro di neuropatia uditiva rilevato in questa categoria di bambini è strettamente correlato alla perdita delle IHCs e quindi a una lesione pre-sinaptica.
A parte la variabilità del danno uditivo periferico, un ulteriore elemento che contribuisce in maniera consistente alla complessità del quadro diagnostico in questi pazienti è correlato con le alterazioni del livello di sincronizzazione dei generatori centrali dell’ABR, anch’esso dipendente dall’effetto dei fattori di rischio che agiscono nella TIN, tra i quali si annoverano, in particolare, la prematurità e l’ipossia in tutte le sue varianti (Kraus et al, 1984; Jiang et al, 2008), Le alterazioni dell’ABR, pertanto, potrebbero risultare dell’effetto combinato della lesione periferica nelle sue varie forme, e dalla ridotta sincronizzazione delle reti centrali con la pratica impossibilità della distinzione del peso relativo di ciascuna componente con l’utilizzo dei procedimenti diagnostici consuetamente utilizzati.
Neuropatia Uditiva e Potenziali cocleari
Almeno in prima approssimazione, la diagnosi di neuropatia uditiva non presenta particolari difficoltà in pazienti che mostrano un quadro uditivo tipico, soprattutto se associato a una polineuropatia periferica correlata con una diagnosi genetica definita. Questa affermazione è sostanzialmente valida anche per le forme di AN sottese dalle mutazioni di otoferlina che comportano l’inattivazione della proteina. Tuttavia, è documentato che le OAEs scompaiono in almeno un terzo dei pazienti che mostrano un tipico quadro di AN (Starr et al, 2008; Rodriguez-Ballesteros et ai, 2008). Inoltre, in alcuni soggetti la soglia uditiva può presentarsi normale (Butinar et al, 2008), mentre in altri casi la percezione verbale risulta compromessa solo in particolari condizioni come l’aumento della temperatura corporea (Wynne et al., 2013) o in presenza di rumore di competizione (Butinar et al, 2008; Starr et al, 2008). In tutte queste situazioni, ai fini di un corretto inquadramento diagnostico, si rende necessario il ricorso a una valutazione obiettiva specifica che dimostri Ici presenza di una alterazione della funzione uditiva periferica. Questo obiettivo può essere conseguito unicamente attraverso la valutazione della morfologia e dei parametri che caratterizzano i potenziali cocleari registrati mediante elettrococleografia con tecnica intratimpanica (electrocochleography, ECochG) ( McMahon et al, 2008; Santarelli et al, 2008). L’indicazione diventa evidentemente anche più rilevante nei bambini dimessi dalla TINI per la possibile sovrapposizione di una componente di desincronizzazione centrale che contribuisce in misura variabile e imprevedibile alle alterazioni della risposta ABR (Santarelli e Arslan, 2013),
Più in generale, tutte le forme di neuropatia uditiva, incluse quelle che non sembrano apparentemente mostrare specifiche difficoltà diagnostiche, richiedono comunque il ricorso a una definizione delle caratteristiche dei potenziali generati perifericamente. Questa valutazione si rende necessaria al fine di stabilire il sito di lesione, che potrebbe essere correlato a una disfunzione delle IHCs o a un disordine primitivamente neurale che coinvolge le fibre del nervo uditivo. Tale informazione è ovviamente cruciale ai fini della programmazione della strategia terapeutica per quel che riguarda in particolar modo la formulazione di una previsione riguardo ai benefici dell’utilizzo dell’impianto cocleare.
Lesioni delle IHCs e del nervo uditivo
La risposta registrata all’elettrococleografia risulta dalla sovrapposizione di tre componenti, delle quali due sono di origine recettoriale, il microfonico coclea
re (cochlear microphonic, CM) e il potenziale di sommazione (summating potential, SP), mentre l’altra risulta dall’attivazione sincrona delle fibre del nervo uditivo (compound action potential, CAP) (Eggermont, 1974; Santarelli e Arslan, 2013). Il microfonico cocleare origina dalla somma delle componenti extracellulari dei potenziali di recettore che si generano per l’attivazione delle OHCs (Eggermont, 1974), mentre il potenziale di sommazione registrato al promontorio è correlato con la componente di distorsione del potenziale di recettore recettoriale relativa all’attivazione delle IHCs localizzate nella porzione basale della partizione coclea-re (Durrant et al, 1998).
Un esempio di registrazione ottenuto da un orecchio normoudente durante la stimolazione con clicks presentati a intensità decrescenti di stimolazione è riportato nella Fig. 2, il microfonico cocleare appare come una attività con andamento oscillatorio che si sovrappone al complesso SP-CAP. Le risposte mostrate nella figura sono state ottenute dopo aver effettuato una preliminare elaborazione dei potenziali registrati alle varie intensità. Infatti, dal momento che il microfonico cocleare riproduce strettamente lo spostamento della membrana basilare, è possibile effettuarne la cancellazione con
|
|
Fig. 2. Potenziali cocleari ottenuti mediante registrazione dell’Eleffrococleografla (ECochG) a intensità decrescenti di stimolazione in un bambino normoudente. |

conseguente estrazione delle varie componenti della risposta ECochG utilizzando un procedimento che consiste nel mediare i potenziali evocati separatamente dalla presentazione di stimoli in condensazione o rarefazione (Eggermont, 1974). Nel soggetto normoudente la risposta ottenuta dopo la cancellazione del CM inizia con una rapida deflessione di polarità negativa, l’SP, che si inscrive quindi nella fase iniziale della risposta neurale. Questultima è rappresentata da un picco negativo con ritorno alla baseline entro 1.5-2 ms alle elevate intensità di stimolazione. Entrambe le componenti, SP e CAP; si riducono di ampiezza e mostrano un aumento della latenza di picco al ridursi dell’intensità dello stimolo, mentre la durata dell’intero complesso SP-CAP mostra un significativo aumento solo in prossimità della soglia (Eggermont, 1974; Santareili e Arslan, 2013).
Le caratteristiche della risposta ECochG risultano alterate profondamente e in vario grado nelle diverse forme di neuropatia uditiva. Un tipico esempio di registrazione ottenuto da una paziente affetta da atrofia ottica dominante per la presenza della mutazione R445H in OPA1 è riportato nella Fig. 3.
Le risposte registrate a intensità decrescente di stimolazione sono state sovrapposte alle corrispondenti curve registrate da un soggetto normoudente. Come si può rilevare, le risposte ottenute dal soggetto con AN appaiono come un potenziale negativo di ampiezza ridotta nel quale non è possibile distinguere separatamente le componenti SP e CAP. Inoltre, rispetto al normale, tale risposta mostra un ritardo della latenza di picco e un considerevole aumento di durata (Huang et al, 2009),
|
|
Fig. 3. Potenziali cocleari ottenuti mediante registrazione dell’Eleffrococleografla (ECochG) in una paziente che presento a mutazione R445H in OPA1. Le risposte registrate a intensità decrescenti di stimolazione sono state sovrapposte ai corrispondenti tracciati ottenuti da un soggetto normoudente. |

Al fine di stabile la natura di tale risposta, se cioè essa originasse da una componente neurale o recettoriale, è stato utilizzato un protocollo di adattamento neurale che consiste nella presentazione di un click iniziale seguito a distanza di 15 ms da un treno di 10 clicks separati da un intervallo di 2.9 ms. Questa sequenza veniva ripetuta ogni 191 ms. Le risposte ottenute nella paziente con AN alla intensità di 110 dB SPL sono state messe a confronto con le corrispondenti risposte ottenute da un soggetto normoudente nella Fig. 4.
|
|
Fig. 4. Adattamento dei potenziali cocleari ottenuti mediante registrazione dell’elettrococleografia (ECochG)alla intensità di 110 dB SPL in risposta al paradigma di stimolazione riportato nella parte inferiore della figura. I tracciati si riferiscono rispettivamente a un soggetto normoudente, una paziente con mutazione R445H in OPA1, un bambino con mutazione biallelica in OTOF e un bambino con il tipico quadro elettrofisiologico di neuropatia uditiva proveniente dalla TIN
|

Nel soggetto normale l’ampiezza del CAP si riduce considerevolmente dai primo al secondo stimolo della sequenza e per poi mostrare un’ulteriore riduzione nel corso dei primi 3-4 clicks del treno ad alta frequenza. Di conseguenza l’ampiezza della risposta, misurata dalli baseline al picco, si riduce del 47% dal primo all’ultimo stimolo della sequenza (media calcolata per 34 orecchie di soggetti normoudenti 57%, range 41-75%)(Sarytarelli et al, 2008; Santarelli e Arslan, 2013). Anche l’ampiezza dell’SP risulta attenuata durante la stimolazione ad alta frequenza, ma il grado di attenuazione (22%) è di gran lunga inferiore rispetto a quello calcolato per il CAP (media calcolata per 34 orecchie di soggetti normoudenti 27%, range 0-53%). Nel soggetto con AN le variazioni di ampiezza sono state misurate unicamente sulla risposta nel suo complesso, non essendo possibile la distinzione tra SP e CAP. Anche in questo caso si osserva una attenuazione della risposta dal primo al secondo click della sequenza seguita da un’ulteriore riduzione durante la stimolazione con treni di click. li grado di attenuazione totale è risultato del 50%, e quindi dello stesso ordine di grandezza della riduzione calcolata per la componente CAP nel normoudente. Si conclude quindi che il potenziale di bassa ampiezza e lunga durata osservato in questa categoria di pazienti con AN deriva verosimilmente dall’anomala attivazione delle fibre nervose.
Analoghe risposte sono state registrate in pazienti con AN sostenute da eziologia di altro tipo, come per es. soggetti affetti da polineuropatia tipo Charcot-Marie-Tooth o disordini neurologici fin qui non categorizzati (Santarelli e Arslan, 2013).
In tutti questi casi la riduzione del numero degli assoni e í fenomeni di demielinizzazione a carico dei neuroni residui costituiscono una ragionevole base per la spiegazione della riduzione di ampiezza e l’aumento di durata delle risposte neurali. Tuttavia, la mancata identificazione dell’SP in alcune neuropatie, come quelle dovute alla mutazione in OPAI, potrebbe indicare un coinvolgimento primario o coesistente delle IHCs con la patologia neurale. Questo rappresenta un aspetto rilevante dal punto di vista del trattamento riabilitativo in quanto l’efficacia della stimolazione elettrica ottenuta con un impianto cocleare dipende essenzialmente dal numero e dalla funzionalità delle fibre neurali residue nelle forme a prevalente coinvolgimento neurale, mentre è indipendente da tale fattore nelle condizioni in cui la neuropatia sia sostenuta unicamente dalla perdita della componente recettoriale.
Disordini delle sinapsi a nastro: alterazioni della funzione dell’otoferlina
Nei bambini con mutazione biailelica in OTOF la risposta ECochG mostra un CM di ampiezza normale (Santarelli et al, 2009). Questo dato è in sostanziale accordo con la conservazione della funzionalità delle OHCs Indicata dalla presenza delle otoemissioni acustiche. Dopo la cancellazione del CM la risposta ECochG mostra la presenza del potenziale di bassa ampiezza e lunga durata registrato in altre forme di AN (Santarelli et al, 2009). In particolare, le risposte registrate all’intensità di 120 dB SPL da cinque bambini con mutazione biallelica in OTOF, che presentavano una ipoacusia profonda associata alla presenza delle otoemissioni acustiche, sono riportate nella Fig. 5 (lato sinistro). I tracciati sono stati sovrapposti al “Grand Average” ottenuto alla stessa intensità da un gruppo di 26 bambini normoudenti. Come si può rilevare dalla figura, le risposte ottenute dai piccoli pazienti iniziano con una rapida deflessione negativa, verosimilmente identificabile come SP per la sua sovrapposizione in termini di ampiezza e latenza con il potenziale sommazione registrato nel normale. Questo dato è indicativo di una normale attivazione delle IHCs nei piccoli pazienti con mutazioni nel gene codificante per otoferlina.
L’SP è seguito da un potenziale di bassa ampiezza e lunga durata al quale si sovrappone in alcuni casi un CAP di ampiezza bassissima. E’ interessante rilevare
come la componente prolungata sia identificabile a intensità ben inferiori rispetto alla soglia psicoacustica (Fig. 5, lato destro). Anche nelle neuropatie dovute ad una alterata funzione dell’otoferlina la risposta di lunga durata che segue l’SP è ragionevolmente generato a livello della fibra nervosa, dal momento che il grado di attenuazione rilevato con l’utilizzo di un protocollo di adattamento
|
|
|

Fig. 5. Potenziali cocleari ottenuti mediante registrazione dell’elettrococleografia (ECochG) in bambini portatori di mutazione biallelica in OMR Nella parte sinistra le risposte registrate a 120 dB SPL in cinque soggetti sono state sovrapposte al Grand Average dei tracciati corrispondenti ottenuti da 26 bambini normoudenti. Le risposte registrate da un paziente a intensità decrescenti di stimolazione sono illustrate nella parte destra della figura.
neurale è dello stesso ordine di grandezza (47%) di quello rilevato per il CAP nel soggetto normoudente (Fig. 4).
Il tipo di risposta registrato nei bambini con mutazione biallelica in OTOF è simile a quello ottenuto da Patuzzi e collaboratori (Sellick et al, 2003) nell’animale da esperimento dopo blocco della generazione dei potenziali d’azione per somministrazione intratimpanica di tetrodotossina. Si potrebbe pertanto ragionevolmente ipotizzare che l’assenza della funzione dell’otoferlina determini una alterazione del rilascio sinaptico nei termini di una riduzione della quota di neurotrasmettitore rilasciato associata alla compromissione del rilascio multivescicolare. Ne consegue una ridotta attivazione della componente dendritica della fibra nervosa con generazione di potenziali post-sinaptici ridotti di ampiezza e dispersi nel tempo, i quali solo occasionalmente e alle alte intensità raggiungono il valore di soglia per l’innesco del potenziale di azione. In ultima analisi, il potenziale di lunga durata registrato con l’elettrococleografia potrebbe derivare dalla somma della componente extracellulare di tutti questi eventi “sotto-soglia”. Pertanto, i dati forniti dalla registrazione dell’ECochG presentano notevoli implicazioni dal punto di vista clinico-terapeutico dai momento che, confermando le ipotesi relative alla localizzazione e al meccanismo del danno formulate in ambito sperimentale, inducono a ritenere che le fibre nervose “per se” non sono coinvolte dal processo patologico. Su questa base è possibile spiegare l’eccellente outcome dell’impianto cocleare osservato in tutti i pazienti affetti da questo tipo di disordine (Rouillon et al, 2006; Rodríguez-Ballesteros et al, 2008; Santarelli et al, 2011).
Potenziali cocleari registrati dai bambini dimessi dalla TIN: lesione delle IHCs o disordine neurale?
Uno dei criteri fondamentali nella identificazione di un quadro di neuropatia uditiva è la riduzione della percezione verbale a fronte di una ipoacusia di grado lieve o moderato, Nei piccoli pazienti selezionati con procedure di screening evidentemente questo tipo di valutazione non può essere effettuato e pertanto l’unico segno distintivo è rappresentato dal riscontro OAEs associato all’assenza della risposta ABR. In questi casi ovviamente solo la registrazione dell’ECochG può fornire informazioni sulla funzione della periferici uditiva per quel che riguarda sia la componente neurale sia i potenziali recettoriali. In aggiunta, i bambini provenienti dalla TIN possono presentare una desincronizzazione dell’attività elettrica dei generatori centrali con conseguente dissociazione tra la soglia della risposta ABR e rispettivamente la soglia uditiva determinata con metodo psicoacustico (Kraus et al, 1984) e la soglia della risposta neurale registrata all’ ECochG (Arslan et al, 1997).
Focalizzando l’analisi ai bambini dimessi dalla TIN che presentano il tipico quadro elettrofisiologico della neuropatia uditiva, vengono riportate alcune osservazioni relative a 23 bambini sottoposti alla registrazione dell’ECochG presso il nostro Servizio dal 2001 al 2012 (età media 23.6 mesi). l potenziali cocleari registrati a livelli
|
|
Fig, 6. Potenziali cocleari ottenuti mediante registrazione dell’elettrococleografia (ECochG) in un bambino con il tipico quadro elettrofisiologico di neuropatia uditiva proveniente dalla TIN. Le risposte registrate a intensità decrescenti di stimolazione sono state sovrapposte ai corrispondenti tracciati ottenuti da un soggetto normoudente. |

nute da un soggetto normoudente nella Fig. 6. La risposta registrata nel soggetto patologico inizia con un potenziale di sommazione, di ampiezza ridotta rispetto al normale, seguito dalla componente di lunga durata analoga a quella registrata in altre forme di neuropatia. L’utilizzo di un protocollo di adattamento (Fig. 4) conferma la genesi neurale di tale componente, dal momento che la riduzione di ampiezza al termine della stimolazione ad alta frequenza risulta pari al 60%.in questo particolare bambino la riduzione di ampiezza del potenziale di sommazione indicativa di un difetto ci attivazione delle IHCs. Questo dato sarebbe pertanto in accordo con il reperto di una riduzione selettiva delle IHCs evidenziato all’esame istologico post-mortem nel gruppo di piccoli pazienti deceduti in TIN che sono studiati da Liberman e collaboratori (Amatuzzi et al, 2011). Tuttavia, oltreché alla riduzione del numero di IHCs, un’alterata attivazione delle fibre nervose potrebbe risultare anche da un coinvolgimento patologico della componente dendritica delle fibre stesse, analogamente a quanto evidenziato in animali da esperimento in seguito all’induzione di un trauma acustico(Lin et al, 2011). E’ pertanto necessario approfondire lo studio dei potenziali cocleari in questa categoria di pazienti e correlarlo con i dati forniti da altre indagini strumentali al fine di chiarire il processo fisiopatologico alla base del disordine uditivo.
Inaspettatamente, risposte di lunga durata sono state registrati anche in bambini (59 soggetti) provenienti dalla TIN che mostravano alterazioni della risposta ABR associate all’assenza di OAEs. In particolare, il 61% di questi soggetti presentava il quadro tipico di una risposta prolungata con assenza di distinzione tra SP e CAP, nel 13% l’SP era identificabile ed era seguito da una risposta neurale di durata aumentata, mentre nel restante 25% i potenziali cocleari erano assenti ad eccezione del CM. In altri termini, il quadro elettrofisiologico riscontrato nella maggioranza di questi soggetti è sovrapponibile a quello rilevato nei pazienti che mostrano il profilo elettrofisiologico della neuropatia uditiva, mentre solo un quarto dei soggetti è risultato affetto da una ipoacusia di grado severo-profondo. Tenendo conto della variabilità dei fattori di rischio che agiscono nella TIN, è possibile ipotizzare che i relativi effetti si traducano in uno “spettro” di lesioni cocleari che comprendono in varia misura la perdita di cellule ciliate interne ed esterne, li danno sinaptico e la sofferenza delle fibre nervose. In questa ottica, ciò che è veramente importante ai fini della diagnosi e della programmazione della strategia terapeutica non è tanto l’associazione ABR assente-OAEs presenti, quanto piuttosto la valutazione della localizzazione e del tipo di danno che coinvolge la periferia uditiva,
Conclusioni
La registrazione dei potenziali cocleari con l’utilizzo dell’elettrococleografia intratimpanica permette di evidenziare alterazioni dei potenziali ,recettoriali e della risposta neurale nei pazienti che presentano un quadro di neuropatia uditiva. In particolare, l’esecuzione dì questo esame permette di definire la diagnosi e di ipotizzare possibili meccanismi fisiopatologici alla base dell’alterazione della funzione neurale. Tali informazioni sono cruciali nella programmazione della strategia terapeutica per quel che riguarda in particolare l’indicazione all’utilizzo di un impianto cocleare per ripristinare la codifica temporale dell’ingresso acustico.
Ringraziamenti
I ringraziamenti sono rivolti a tutto ii personale del Servizio di Audiologia e Foniatria che ha contribuito fattivamente alla valutazione clinica dei pazienti con neuropatia uditiva e allo sviluppo della ricerca centrata su questo tema. Gli autori ringraziano in particolare la Dr.ssa Roberta Rossi, il Dr. Pietro Scimemi e la Dr.ssa Erica Dal Monte per il loro contributo alla raccolta e alla analisi dei dati. Un grazie sentito deve essere rivolto inoltre agii anestesisti e agli infermieri della sala operatoria e alla ULSS9, che ha finanziato in gran parte il laboratorio di elettrofisiologia. Infine, un ringraziamento particolare deve essere rivolto agli amici e colleghi collaboratori appartenenti ad altre università: Prof. Arnold Starr, Professor of Neurology at University of California lrvine; Prof. Ignacio del Costino, Unidad de Genéticci Molecular dell’Hospital Ramón y Cajal di Madrid; Prof. Valerio Carelli, Dipartimento di Science Neurologiche dell’Università di Bologna.
Questo lavoro è dedicato alla memoria del prof. Arslan: la storia della nostra ricerca insieme.
INDIRIZZO PER CORRISPONDENZA: DR.SSA ROSAMARIA SANTAREW, DIPARTIMENTO DI NEUROSCIENZE, STRUTTURA COMPLESSA DI AUDIOLOGIA E FONIATRIA, UNIVERSITÀ DI PADOVA, ULSS 9 DI TREVISO,
TEL, (+39) 0422 328286: FAX: (+39) 0422 322351; E-MAIL: ROSAMARIA.SANTAREW@UNIPD.IT
Bibliografia
Amatuzzi M, Liberman MC, Northrop C. Selective inner hair cell loss in prematurity: a temporal bone study of infants from a neonatal intensive care unit, J. Assoc. Res, Otolaryngol. 2011; 12: 595-604.
American Academy of Pediatrics, Joint Committee on Infant Hearing. Year 2007 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics 2007; 120: 898-921.
Arslan E, Turrini M, Lupi G et ai. Hearing threshold assessment with auditory brainstem response (A8R) and ElectroCochleoGraphy (ECochG) in uncooperative children. Scand Audiol Suppl 1997; 46: 32-37,
Berlin Cl, Hood LJ, Morlet T et al. Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder). Int J Audiol 2010; 49: 30-43,
Butinar D, Starr A, Zidar J et al. Auditory nerve is affected in one of twa different paint mutations of the neurofilament light gene. ain Neurophysiol 2008; 119: 367-375.
Carelli V, boss-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004; 23: 53-89.
Delettre C, Lenaers G, Griffoin JM et ai. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000; 26: 207-210.
Durrant JD, Wang J, Ding DL et al. Are inner or outer hair cells the source of summating potentials recorded from the round window? J, Acoust. Soc, Am. 1998; 104: 370-377.
Eggermont JJ. Basic principles for electrocochleography, Acta Otolaryngol Suppl 1974; 316: 7-16,
Frezza C, Cipolat S, Martins de Brito O et al. OPA1 controls apoptotic crista.0000e remodeling independently from mitochondrial fusion. Cell 2006; 126: 177-189.
Hille ETM. van Straaten HI, Verkerk PH et al. Prevalence and independent risk factors for hearing boss in NICU infants. Acta Paediatr. 2007; 96: 1155-1158.
Huang T Santarelli R, Starr A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res. 2009; 1300: 97-104.
Jiang ZD, Brosi DM, Shao XM et al. Sustained depression of brainstemn auditory electrophysiology during the first months in term infants after perinatal! asphyxia. Clin Neurophysiol 2008; 119: 1496-1505.
Johnson JS, Newport EL. Critical period effects on universal properties of language: the status of subjacency in the acquisition of a second language. Cognition 1991; 39: 215-258.
Kaga K, Starr A Neuropathies of the Auditory and Vestibular Eighth Cranial Nerves. Springer; 2009.
Kjer P. Infantile optic atrophy with dominant mode af inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol. Suppl. 1959; 164: 1-147.
Kovach MJ, Campbell KCM, Herman K et al. Anticipation in a unique family with Charcot-Marie-Tooth syndrome and deafness: delineation of the clinical features and review of the literature. Am. J. Med. Genet. 2002; 108: 295-303.
Kraus N, Ozdamar O, Stein L et al. Absent auditory brain stem response: peripheral hearing loss or brain stem dysfunction? Laryngoscope 1984; 94: 400-406.
Lin HW, Furman AC, Kujawa 5G et al. Primary neural degeneration in the Guinea pig cochlea affer reversible noise-induced threshold shift. J. Assoc. Res, Otolaryngol. 2011; 12: 605-616.
McMahon CM, Patuzzi RB, Gibson WPR et ai. Frequency-specific electrocochleography indicates that presynaptic and postsynaptic mechanisms of auditory neuropathy exist. Ear Hear 2008; 29: 314-325.
Olichon A, Baricault L, Gas N et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome e release and apoptosis. J. Biol. Chem. 2003; 278: 7743-7746.
Pangrsic T Lasarow L, Reuter K et al. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Net. Neurosci. 2010; 13: 869-876.
Rizo J, Sűdhof TC. C2-domains, structure and function of a universal Ca2+ binding domain. J. Biol. Chem. 1998; 273: 15879-15882.
Rodriguez-Ballesteros M, del Castillo FJ, Martin Y et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum. Mutat. 2003; 22: 451-456.
Rodriguez-Ballesteros M, Reynosa R, Olarte M et al. A multicenter study on the prevalence and spectrum af mutations in the otoferlin gene (OTOF) in subjects with non-sindromic hearing impairment and auditory neuropathy. Hum. Mutat, 2008; 29: 823-831.
Rouillon I, Marcolla A, Roux I et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int. J. Pediatr. Otorhinolaryngol. 2006; 70: 689-696.
Roux I, Safieddine S, Nouvicin R et al. Otoferlin, defective in a human deafness form, is essential for exocytosis al the auditory ribbon synapse. Cell 2006; 127: 277-289.
Santarelli R, Arslan E. Electrocochleography, in: Disorders of Peripheral and Central Auditory Processing. Handbook of Clinica! Neurophysiology, New York: GG Celesia; 2013. p. 83-113.
Santarelli R, Del Castillo l, Rodríguez-Ballesteros M et al. Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. J. Assoc. Res. Otolaryngol. 2009; 10: 545-556.
Santarelli R, Scimemi P, Dal Monte et al Auditory neuropathy in systemic sclerosis: a speech perception and evoked potential study before and after cochlear implantation. Eur Arch Otorhinolaryngol 2006; 263: 809-815.
Santarelli R, Starr A. Michabewski HJ et al. Neural and receptor cochlear potentials obtained by Transtympanic electrocochleography in auditory neuropathy. Clin Neurophysiol 2008; 119: 1028-1041.
Santarelli R. Information from cochlear potentials and genetic mutations helps localize the lesion site in auditory neuropathy. Genome med 2010:2:91
Santarelli R. Starr A, del Castillo I et al. Presynaptic and postsynaptic mechanisms underlying auditory neuropathy in patients with mutations in the OTOF or OPA 1 gene. Audiological Medicine 2011; 9: 59-66.
Sellick P, Patuzzi R, Robertson D. Primary afferent and cochlear nucleus contributions to extracellular potentials during Ione-bursts. Hear. Res. 2003; 176: 42-58.
Singer JH, Glowatzki E. Moser T et al. Functional properties of synaptic transmission in primary sense organs. J. Neurosci. 2009; 29: 12802-12806,
Starr A, Michalewski HJ, Zeng FG ef al. Pathology and phycology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145->Ser), Brain 2003; 126: 1604-1619,
Starr A, Picton 1W .Sininger Y et al. Auditory neuropathy. Brain 1996; 119 ( Pt 3): 741-753.
Starr A, Sininger YS, Pratt H. The varieties of auditory neuropathy. J Basic Clin Physiol Pharmacol 2000: 11: 215-230.
Starr A, Zeng E Michalewski H et al. Perspectives on Auditory Neuropathy: Disorders of Inner Hair Cell, Auditory Nerve, and Their Synapse (Internet). In: Volume 3: Audition. Elsevier; 2008. p. 397-412.
Varga R, Avenarus MR, Kelley PM et al. OTOF mutations revealed by genetic analysis of hearing loss families including o potential temperature sensitive auditory neuropathy allele. J. Med. Genet, 2006; 43: 576-581.
Varga R, Kelley PM, Keats BJ et al. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J. Med. Genet, 2003; 40: 45-50.
Wang D-Y, Wang V-C, Weil D et al. Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med. Genet. 2010; 11: 79.
Wynne DP, Zeng F-G, Bhatt S et al. Loudness adaptation accompanying
ribbon synapse and auditory nerve disorders. Brain 2013; 136: 1626-1638.
Xoinis K, Weirather Y, Mavoori H et al. Extremely low birth weight infants ore at high risk for auditory neuropathy. J Perinatol 2007; 27: 718-723.
Zeng FG, Kong YY, Michalewski HJ ef al. Perceptual consequences of disrupted auditory nerve activity. J. Neurophysiol, 2005; 93: 3050-30
|
CASO CLINICO |
|
|
Neuropatia uditiva / dissincronia uditiva – una condizione sottodiagnosticata: un caso clinico con revisione della letteratura
Vinish Agarwal,Saurabh Varshney,Sampan Singh Bist,Sanjiv Bhagat,Sarita Mishra,Vivek Jha
Dipartimento di ORL, Himalayan Institute of Medical Sciences, (hiht University), Jollygrant, Doiwala, Dehradun, Uttaranchal, India
|
Data di pubblicazione Web |
12-Nov-2012 |
|
|
Corrispondenza Indirizzo:
Saurabh Varshney
Dipartimento di ORL, Himalayan Institute of Medical Sciences, (hiht University), Jollygrant, Doiwala, Dehradun 248 140, Uttaranchal
India
![]()
Fonte di sostegno: nessuno, Conflitto di Interessi: Nessuno
DOI: 10,4103 / 0971-7.749,103445
![]()
Neuropatia uditiva (AN) / dissincronia uditiva (AD) è una diagnosi molto spesso perso, quindi una condizione sottodiagnosticata nella pratica clinica. Neuropatia uditiva è una condizione in cui i pazienti, su una valutazione audiologica, si trovano ad avere la normale funzione delle cellule ciliate esterne e la funzione neuronale anormale a livello di ottavo nervo. Questi pazienti, sui test clinici, si trovano ad avere normali otoemissioni acustiche, mentre uditivi del tronco encefalico audiometria risposta rivela l’assenza di sincronia neurale. A differenza di lesioni occupanti spazio, valutazione radiologica rivela risultati normali. I pazienti con neuropatia uditiva richiedono un approccio di gestione diverso ai loro problemi uditivi e di comunicazione da approcci utilizzati con i pazienti con normali perdite uditive periferiche.
Parole chiave: risposte uditivi del tronco encefalico, neuropatia uditiva, emissione Otoacoustic
|
Come citare questo articolo: |
|
Come citare questo URL: |
|
|
Neuropatia uditiva (AN), noto anche come ‘neurale dissincronia / dissincronia uditiva (AD), [1] è una perdita di udito a causa di processi neurali anomali di stimoli uditivi. I pazienti affetti da questa malattia sono in grado di rispondere in modo appropriato ai suoni, ma la loro capacità di decodificare parola e del linguaggio è ostacolato. Questi pazienti sono perdite che vanno da lieve a grave con scarse capacità di percezione del linguaggio udito. AN / AD è stato recentemente descritto soltanto. Alla fine del 1970, ricercatori clinici hanno cominciato a descrivere gruppi di pazienti con audiogramma soglie normali o leggermente elevati di tono puro accompagnati con risposte uditiva del tronco encefalico assenti o gravemente anormali (ABRs). Con l’avvento delle otoemissioni acustiche (OAEs) a metà degli anni 1980, questi gruppi di pazienti sono stati trovati ad avere la normale funzione cocleare. La scoperta della normale funzione cocleare accompagnato con le risposte del tronco encefalico anomale è stato definito nel 1996 come neuropatia uditiva (AN). Se questo rappresenta un vero neuropatia nervo acustico è discutibile. Ulteriori indagini hanno portato alla conclusione che AN può veramente rappresentare un nervo uditivo dyssynchronous piuttosto che una neuropatia. Questa scoperta ha dato origine a una nuova durata del dissincronia uditiva (AD). [1] Ai fini del presente articolo, AN e AD sono considerati sinonimi (cioè, AN / AD). AN può colpire persone di ogni età, dall’infanzia all’età adulta. Lo scopo di questo articolo è quello di discutere presentazione clinica, esami audiologici, e le sue opzioni di gestione, come è molto spesso mancato come diagnosi nella pratica clinica.
|
|
A 16 anni studentessa presentato con lamenta insidioso, perdita progressiva dell’udito per 2 anni in entrambe le orecchie. Non c’era storia di qualsiasi scarico orecchio, traumi, ototossicità, o qualsiasi altra malattia medica o chirurgica. Non c’era prenatale contributivo, perinatale e postnatale la storia. E ‘nata come normale parto vaginale a termine in ospedale. Non c’erano storia suggestiva di perdita di udito in famiglia.All’esame clinico, otoscopia bilaterale era normale. In esecuzione di test diapason, il test di Rinne è stato positivo a livello bilaterale, prova di Weber era centralizzata, e test di conduzione ossea assoluto è stato ridotto bilateralmente. Resto dell’esame ORL e testa collo era normale.Audiometria tonale (PTA) ha mostrato moderatamente grave ipoacusia neurosensoriale (Med. 65 dB) sul lato destro e da lieve a moderata ipoacusia neurosensoriale (Med. 40 dB) sul lato sinistro, che era più per le frequenze più basse [Figura 1]. Punteggi di discriminazione Discorso erano fuori proporzione con sospetta ipoacusia. Timpanometria mostrato bilateralmente ‘A’ tipo di curva, con assenza di riflessi ipsilaterale e controlaterale [Figura 2]. Otoemissioni acustiche (DPOAE / TPOAE) erano presenti per entrambe le orecchie [Figura 3]. Su ABR, senza V definitiva th onde sono stati visti su entrambi i lati, anche dopo 90 dB e 96 dB [Figura 4]. Il paziente è stato sottoposto a risonanza magnetica per immagini (MRI) del cervello e HRTC osso temporale, che erano normali. Quindi, una diagnosi di AN è stato fatto come paziente aveva storia della perdita di udito bilaterale; PTA ha mostrato sordità neurosensoriale bilaterale, OAE era presente bilateralmente, ma ABR mostrava grave ipoacusia neurosensoriale in entrambe le orecchie. Il paziente è stato consigliato impianto cocleare, ma non poteva permetterselo. Quindi, il paziente è stato consigliato l’uso di un aiuto per entrambe le orecchie, che lei sta usando con un certo guadagno udito.
|
|
Figura 1: Pure Tone Audiogram- Ear-moderata Diritto di perdita uditiva severa SN; Ear-sinistra da lieve a moderata perdita di udito SN. (Nota: componente conduttivo potrebbe essere visto in bassa frequenza) |

|
|
Figura 2: Tympanometry- bilaterale di tipo “A” di curva, senza ipsi e controlaterale riflesso acustico.(Impression: Nessuna indicazione di patologia dell’orecchio medio) |

|
|
Figura 3 (a, b): emissione Otoacostic (OAE) – TEAOE e DPOAE- mostrando funzione bilaterale normale cocleare |

|
|
Figura 4: ABR- uditiva del cervello response- non definita onda V sono stati visti su entrambi i lati, anche dopo 90 dB e 96 dB. |

Fisiopatologia
Il termine neuropatia uditiva / dissincronia uditiva (AN / AD) descrive una diagnosi che colpisce un piccolo gruppo di pazienti con problemi di udito punteggi perdita e intelligibilità del parlato fuori proporzione con la loro perdita di udito presunta. Molti autori hanno suggerito che le anomalie che causano un risiedono nel sistema uditivo inferiore. In particolare, le cellule gangliari a spirale, nervo uditivo, oppure i nuclei del tronco cerebrale uditive sono stati tutti coinvolti. La combinazione di un nervo uditivo disfunzionale con conservazione della funzione cocleare può teoricamente essere causato a vari punti lungo il percorso uditivo inferiore. Sono stati proposti i seguenti anomalie:
- Lesioni alla giunzione sinaptica tra le cellule ciliate interne della coclea e dendriti dei neuroni gangliari a spirale
- Danno diretto alle dendriti dei neuroni gangliari spirale
- Danno diretto ai neuroni gangliari spirale
- Danno diretto assonale al nervo uditivo che provoca una cascata di danni al nuclei uditivo più bassa
|
|
Neuropatia uditiva è stata la prima volta nel 1970 in ritardo di come risultati paradossali tra ABR e OAE e è stata definita come la neuropatia uditiva (AN) nel 1996 da Arnold Starr. [2] Madden et al. ha rilevato che 22 su 428 bambini affetti da perdita dell’udito aveva una. [3] Alcuni autori hanno suggerito che la prevalenza è 2-15% dei bambini con perdita uditiva nota. In una revisione della prevalenza 2002 Sininger suggerito che circa 1 su 10 bambini affetti da perdita e gravemente colpiti i risultati dei test ABR udito hanno AN. Nel complesso, AN è raro e può essere trovato in una stima di 1-3 bambini ogni 10.000 nascite. [4] Nel sud dell’India, la prevalenza di disfunzione uditiva-sincronia è stato segnalato circa 1 183 in individui con ipoacusia sensoriale neurale. [5] neuropatia uditiva / dissincronia uditiva (AN) non ha pregiudizi razziali e si verifica con una frequenza pressoché uguale in maschi e femmine. I pazienti vanno in età da bambino ad adulto e presentano caratteristiche uditive coerenti con la normale funzione delle cellule ciliate esterne e la funzione neuronale anormale a livello dell’VIII ° nervo.Perché AN è una condizione relativamente recente descrizione, molti adulti non possono hanno ottenuto la corretta sperimentazione audiologica per raggiungere una diagnosi di AN. Con l’avvento del neonato test di screening dell’udito, il ritardo nella diagnosi di AN deve essere minimizzata, che accelerare l’intervento.
Cellule gangliari a spirale, nervo uditivo, o nuclei del tronco cerebrale uditive sono stati implicati per le anomalie che causano AN. Due forme di AN sono state suggerite (i) tipo pre-sinaptica e (ii) versione post-sinaptica. [6] AN può anche influenzare la funzione vestibolare. [7] I fattori di rischio per AN includono la storia neonatale di anossia, iperbilirubinemia, ventilazione meccanica , ipossia, anomalie cerebrali congenite, basso peso alla nascita, parto estremamente prematuro (<28 settimana), e la storia genetica e familiare di AN. [3] Berlino et al. descritto AN in tre pazienti della malattia di Charcot-Marrie-Tooth, [8] mentre Doyle, Sininger, e Starr ha riportato AN con atassia di Fredrich. [9] Bamiou et al.descritto AN con la malattia di Refsum. [10] Anche se una complicata storia perinatale è comune tra i più pazienti con AN, un terzo dei pazienti non hanno fattori predisponenti che portano allo sviluppo di AN. Neuropatia uditiva è di solito bilaterale, ma di tanto in tanto può essere unilaterale anche.
Questi sono i criteri per la diagnosi di AN [11]
- I risultati dei test ABR assenti o gravemente anormali a stimoli massima (100 dBnHL)
- Normale funzione delle cellule ciliate esterne, come determinato dalla OAE o CM
- Soglie di riflesso stapediali assenti o elevati
HRTC ossa temporali dovrebbe essere fatto per escludere malformazioni dell’orecchio interno nei neonati. In genere, una risonanza magnetica hanno limitato ruolo in AN (Wang et al. 2003). [12]
Un approccio multidisciplinare tra cui un otologist / neuro-otologist, logopedista, consulente genetico, audiologo, e pediatri sono tenuti a diagnosticare e gestire i pazienti di AN.
Assistenza medica inizia con i genitori e la consulenza includono patologia discorso, sentendo il posizionamento degli aiuti, e l’uso di altri dispositivi di udienza. Recenti studi dimostrano che il 50% dei bambini può essere beneficiato di posizionamento di un dispositivo di amplificazione.
Nel 2001, l’uso di impianto cocleare è stato ampliato per includere i bambini con AN. [13] Più di recente, se l’impianto cocleare non riesce, la possibilità di tronco encefalico impianto è stato riportato. [14]
|
|
Neuropatia uditiva (AN) è un termine usato per descrivere un disturbo uditivo, in cui vi è la prova della funzione delle cellule ciliate esterne normale (otoemissioni acustiche e / o microfonici cocleari) e scarsa funzionalità del nervo uditivo (tronco cerebrale uditivo assente o altamente distorta risposta a partire da onda I). Neuropatia uditiva è un disturbo dell’udito che può colpire qualsiasi età e in gran parte causata da un’anomalia nella bassa percorso uditivo. Può essere diagnosticata da un’attenta storia e valutazione audiologica per OAE e test ABR. Sono a disposizione per far fronte alla situazione varie opzioni mediche e qualche chirurgica. Una certa ripresa nel sistema uditivo è stata riportata, ma la maggior parte dei bambini affetti da AN continuare a visualizzare le medie di toni puri anomale e risultati dei test ABR che richiedono un impegno permanente da parte del bambino, la famiglia, la società, logopedista, e audiologo. In un programma di screening uditivo neonatale, i controlli di ABR e / o microfonici cocleari dovrebbero essere considerati nei neonati ad alto rischio che si sottopongono a test OAE come strumento di screening primario.
|
References |
|
|
Berlin C, Hood L, Rose K. On renaming auditory neuropathy as auditory dyssynchrony. Audiol Today 2001;13:15-7. |
|
|
Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain 1996;119:741-53. |
|
|
Madden C, Rutter M, Hilbert L, Greinwald JH Jr, Choo DI. Clinical and audiological features in auditory neuropathy. Arch Otolaryngol Head Neck Surg 2002;9:1026-30. |
|
|
Sininger YS, Trautwein P. Electrical stimulation of the auditory nerve via cochlear implants in patients with auditory neuropathy. Ann Otol Rhinol Laryngol Suppl 2002;189:29-31. |
|
|
Kumar UA, Jayram MM. Prevalence and audiological characteristics in individuals with auditory neuropathy/auditory dys-synchrony. Int J Audiol 2006;45:360-6. |
|
|
McMahon CM, Patuzzi RB, Gibson WP, Sanli H. Frequency-Specific Electrocochleography Indicates that Presynaptic and Postsynaptic Mechanisms of Auditory Neuropathy Exist. Ear Hear 2008;29:314-25. |
|
|
Sheykholeslami K, Kaga K, Murofushi T, Hughes DW. Vestibular function in auditory neuropathy. Acta Otolaryngol 2000;120:849-54. |
|
|
Berlin CI, Hood LJ, Hurley A, Wen H. Contralateral suppression of otoacoustic emissions: An index of the function of the medial oliovocochlear system. Otolaryngol Head Neck Surg 1994;110:3-21. |
|
|
Doyle KJ, Sininger Y, Starr A. Auditory neuropathy in childhood. laryngoscope 1998;108:1374-7. |
|
|
Bamiou DE, Spraggs PR, Gibberd FB, Sidey MC, Luxon LM. Hearing loss in adult Refsum’s disease. Clin Otolaryngol 2003;28:227-30. |
|
|
Devies RA. Retrocochlear hearing disorder, including auditory dyssynchrony In: Gleeson M, Hilbert JS, editors. Scott- Brown’s Otorhinolaryngology, head and neck surgery. 7th ed, Vol 3. London: Hodder Arnold; table 241a.5, 2008. p. 3842. |
|
|
Wang DY, Bu XK, Xing GQ, Lu L. Neurophysiological characteristics of infants and young children with auditory neuropathy. Zhonghua Yi Xue Za Zhi 2003;83:281-4. |
|
|
Trautwein PG, Sininger YS, Nelson R. Cochlear implantation of auditory neuropathy. J Am Acad Audiol 2000;11:309-15. |
|
|
Colletti V, Fiorino FG, Carner M, Miorelli V, Guida M, Colletti L. Auditory brainstem implant as a salvage treatment after unsuccessful cochlear implantation. Otol Neurotol 2004;25:485-96. |
Neuropatia uditiva: inaspettatamente comune in una popolazione neonatale screening ANDREW C Dowley WILLIAM,, P WHITEHOUSE, STEVE M MASON,YVONNE COPE,JUDITH GRANT,KEVIN P Gibbin Developmental Medicine & Child Neurology Volume 51, Issue 8, pagine 642-646, agosto 2009
Astratto
Neuropatia uditiva, o dissincronia, è definita da una risposta uditivi del tronco encefalico anormale o assente, ma intatto otoemissioni acustiche o microfonici cocleari. Si è associato con problemi di udito in comportamentale audiometria toni puri, dei riflessi acustici assenti, e la scarsa percezione del linguaggio, soprattutto in ambienti rumorosi. Questi risultati suggeriscono un disturbo di capelli-cellulare interna e la funzione o l’ottavo nervo. Descriviamo un sondaggio caso note di pazienti con e senza neuropatia uditiva, utilizzando i dati del programma di screening uditivo neonatale locale raccolti in modo prospettico dal 2002 al 2007. Durante questo periodo, 45 050 bambini sono stati sottoposti a screening con otoemissioni acustiche, 30 pazienti sono stati diagnosticati con sospetta grave di sordità profonda (16 maschi, 14 femmine), e 12 di questi 30 avevano neuropatia uditiva (sei maschi, sei femmine). Età gestazionale media era di 33 settimane 1 giorno nel gruppo neuropatia uditiva e 35 settimane nel gruppo neuropatia non uditiva. I fattori di rischio più significativi per la neuropatia uditiva erano iperbilirubinemia (p = 0.018), sepsi (p = 0,024), e l’esposizione gentamicina (p = 0,024). I bambini con neuropatia uditiva comprendono un sottogruppo di pazienti con insufficienza uditiva che coinvolge diverse patologie più comunemente associati con i fattori di rischio connessi all’ammissione alla unità di terapia intensiva neonatale. Il miglioramento è possibile con la maturità, almeno in una minoranza.
Neuropatia uditiva come entità è stato il primo sospetto nel 1980, quando i pazienti con normali audiogrammi puro-tono presentati con difficoltà di percezione del suono, in particolare in ambienti rumorosi. Con l’avvento del neonato test di screening dell’udito, neuropatia uditiva è stato riconosciuto nella popolazione pediatrica. L’incidenza di neuropatia uditiva nei pazienti con sordità profonda è stimata al 10%, 1,2 con una prevalenza nei bambini a rischio di 0,23%. 2
La definizione di neuropatia uditiva implica normalmente funzionante cellule ciliate cocleari esterno, ma risposte anomale nel percorso dalla coclea al tronco encefalico. Pertanto, questi pazienti hanno normali microfonici cocleari e otoemissioni acustiche, ma le risposte uditiva del tronco encefalico anomali o assenti. Il comportamento audiogramma puro-tono può variare notevolmente, da un udito normale a sordità profonda. Una pendenza inversa (soglie povere frequenze più basse, meglio in alto) è un riscontro comune. I riflessi acustici (ad esempio, di riflesso auropalpebral) sono assenti, e percezione del linguaggio è più povera di quanto sarebbe prevedibile dalle soglie puro-tono comportamentali, in particolare in ambienti rumorosi.
La patologia sottostante varia fra i pazienti, dando diversi quadri clinici, e può essere simile ad altre neuropatie periferiche in alcuni. Il sito della patologia è importante (recettore, sinapsi, o nervo acustico stesso; Fig. 1). Villi interni sono particolarmente sensibili all’ipossia, rispetto alle cellule cigliate esterne, 3 e anche ad alcuni insulti tossici come carboplatino. 4 disturbi sinaptiche possono causare problemi rate-dipendente con saturazione della risposta; per esempio, uno stimolo dato 3 o 11 volte al secondo viene rilevato perfettamente ma è insufficientemente rilevata a 20 volte al secondo. 5 Disturbi del nervo uditivo possono derivare sia da demielinizzazione, che provoca un rallentamento del potenziale d’azione composto, il blocco corrente e trasmissione ephaptic (corrente di dispersione di neuroni adiacenti), o malattia assonale primaria con la perdita di fibre e piccoli potenziali d’azione composto. Questi due disturbi molto probabilmente influenzano i potenziali di azione delle fibre più lunghe (come è il caso nelle neuropatie periferiche) a causa della degenerazione lungo la lunghezza e, come queste fibre forniscono l’apice della coclea, ci si aspetterebbe per causare più impairment in basso frequenze. Ciò è in linea con le osservazioni di cui sopra.

Figura 1. Sezione attraverso la coclea, espanso per mostrare l’organo del Corti.
Nel Regno Unito, la prova delle emissioni otoacustiche è l’unico test di routine eseguiti sulla popolazione neonatale, a meno che non vi sono fattori di rischio significativi per la perdita uditiva profonda, come ad esempio una storia familiare o l’ammissione alla terapia intensiva neonatale, quando viene eseguita anche la risposta uditivi del tronco encefalico. Questo è l’unico gruppo nel popolazione del Regno Unito sia in fase di emissione otoacustiche e test di risposta uditivi del tronco encefalico, quindi è il gruppo predominante in cui neuropatia uditiva è in fase di diagnosi.
Descriviamo un sondaggio caso note dei pazienti con neuropatia uditiva, due dei quali sono stati descritti in un rapporto precedente, 6 e discutere i fattori di rischio coinvolti, rispetto a quelli per i pazienti con diagnosi di grave sordità profonda senza neuropatia uditiva.
Metodo
I dati audiologici del programma locale di screening uditivo neonatale dal suo inizio nel 2002-2007 sono stati raccolti in modo prospettico, come parte di una valutazione del servizio. Le note mediche dei bambini identificati con grave per insufficienza uditiva profonda (60dB soglie) sono stati poi recensione. Questa valutazione non intervento è stato registrato come un audit clinico con il Nottingham University Hospitals NHS Trust e come tale non richiede l’approvazione etica.
Informazioni raccolte audiologico incluse età alla diagnosi (definito come la data in cui i genitori o tutori sono stati informati della diagnosi), età in cui sono stati forniti apparecchi acustici, e l’età di impianto cocleare, se adatto. L’ultima audiogramma comportamentale (rinforzo audiogramma visivo, sia di campo sonoro o l’orecchio specifico) è stato inoltre esaminato se il bambino è stato evolutivo in grado di intraprendere la valutazione.
Le informazioni raccolte dalla fisica medica inclusi i risultati delle otoemissioni acustiche e le risposte uditiva del tronco encefalico. Eravamo particolarmente interessati a sapere se ci fosse stato un segno di maturazione o la prova di un cambiamento di soglia con il tempo. Lo rivela un miglioramento della risposta tronco cerebrale uditivo rispetto test precedenti, come questi bambini sono riesaminati a 3, 9, e 12 mesi di età. 7
La revisione note mediche incluso prenatale dettagliata, la nascita, e storie neonatali, comprese le informazioni sui potenziali fattori di rischio come la gestazione, peso alla nascita, unità di terapia intensiva neonatale ammissione, ipossia, ittero, o sepsi. Per semplicità, tutte le età sono reali e non corretto per la gestazione.
I pazienti con neuropatia uditiva sono stati confrontati con quelli con disabilità uditiva senza neuropatia uditiva, e sono stati identificati i possibili fattori causali.
Statistiche descrittive semplici sono dati, utilizzando il test esatto di Fisher per i valori nominali e accoppiati di Student t-test per i dati di intervallo.
Risultati
Durante lo studio, 45 050 bambini sono stati sottoposti a screening con otoemissioni acustiche, 30 pazienti sono stati diagnosticati con sospetta grave di sordità profonda, e 12 di questi 30 avevano neuropatia uditiva. Questo dà un’incidenza annuale di 0,67 / 1000 per i casi di grave sordità profonda e 0,27 / 1000 per la neuropatia uditiva. Il follow-up medio è stato di 2 anni 10 mesi (range 10mo-5A 1Mo). Tabella I mostra confronto tra i due gruppi.
|
Tabella I. Caratteristiche cliniche di bambini con e senza neuropatia uditiva |
|||
|
Caratteristica clinica |
Neuropatia uditiva |
Neuropatia Non-uditiva |
Significato |
|
|||
|
Numero di pazienti |
12 |
18 |
|
|
Maschi / femmine |
6/6 |
10/8 |
|
|
Deficit uditivo grave |
2 |
4 |
|
|
Menomazione uditiva profonda |
10 |
14 |
|
|
L’ammissione alla terapia intensiva neonatale |
12/12 |
7/18 |
p = 0.001 |
|
Gestazione |
|||
|
Media (DS) |
33wks 1d (32d) |
35wks (43d) |
0,3298 a, p = 0,748 |
|
Gamma |
25wks 2d-40wks |
23wks 0d-40wks |
|
|
Nato a termine |
5/12 |
9/18 |
p = 0,722 |
|
Peso alla nascita, media (SD) g |
1863 (990) |
1976 (1060) |
0,294 a, p = 0,774 |
|
Ventilazione meccanica |
|||
|
Pazienti |
10/12 |
7/18 |
0,9118 a, p = 0,397 |
|
Durata, media (SD) d |
9.2 (9.1) |
34.3 (65.8) |
|
|
L’ipossia |
1/12 |
1/18 |
|
|
Iperbilirubinemia |
4/12 |
0/18 |
p = 0.018 |
|
Emorragia intracranica |
4/12 |
1/18 |
p = 0,128 |
|
Sepsi |
8/12 |
4/18 |
p = 0.024 |
|
Esposizione Gentamicina |
9/12 |
5/18 |
p = 0.024 |
|
La paralisi cerebrale |
2/12 |
1/18 |
p = 0,548 |
I due gruppi erano strettamente comparabili in termini di gestazione, numeri che raggiungono termine, e peso alla nascita. Entrambi i gruppi inclusi alcuni bambini molto bene, ma questo è stato particolarmente evidente nel gruppo di neuropatia uditiva. Come illustrato nella tabella I, tutte le misure di gravità della malattia erano significativamente peggiore per il gruppo neuropatia uditiva rispetto al gruppo non-neuropatia uditiva. Questo ci si aspetta come il gruppo neuropatia non uditiva inclusi quelli con perdita di udito non sindromica che erano spesso ben, e, prima che il programma di screening neonatale, non sarebbe stato diagnosticato problemi di udito fino al più tardi nella vita.
Tutti i bambini con neuropatia uditiva sono stati ricoverati in unità di terapia intensiva neonatale; questo suggerisce che la malattia neonatale, soprattutto associata a parto pretermine (per esempio, l’insufficienza respiratoria che richiede ventilazione meccanica, l’ipossia, la sepsi, iperbilirubinemia, ed emorragia intracranica) svolge un ruolo causale nella neuropatia uditiva, anche se il campione è stato prevenuto nei confronti di coloro con i fattori di rischio neonatali. Ciò è dimostrato anche dal fatto che i più significativi fattori di rischio per la neuropatia uditiva in questo gruppo sono stati iperbilirubinemia (p = 0,018), sepsi (p = 0,024), e l’esposizione gentamicina (p = 0,024).
Tra i bambini che sono stati ventilati, quelli con neuropatia uditiva non sono state ventilate in media più a lungo; tuttavia, questo gruppo comprendeva un outlier con ventilazione totale di 6 mesi. Se questo bambino è scontato, quindi il periodo medio di ventilazione per il gruppo la neuropatia non uditiva era 10,3 giorni, che non era significativamente diverso dal gruppo di neuropatia uditiva (Tabella I). Altri fattori di rischio riportati in precedenza, quali l’ipossia e paralisi cerebrale (CP), non sono stati più frequenti nel gruppo neuropatia uditiva che nel gruppo neuropatia non uditiva nella nostra serie di neonati a rischio.
Il tempo medio per la diagnosi è stata di 3 mesi a 25 giorni per il gruppo uditiva neuropatia (gamma-5d 9mo 26d) e 3 mesi 15 giorni per il gruppo la neuropatia non uditiva (intervallo 28d-25d 8mo). Tutti i pazienti hanno ricevuto apparecchi acustici, e il tempo medio per il primo utilizzo di un apparecchio acustico è stato 4 mesi 27 giorni per il gruppo neuropatia uditiva (gamma 1mo 7d 28d-8mo) e 6 mesi 1 giorno per il gruppo la neuropatia non uditiva (gamma 1Mo 9d-1A 11mo 12d).
Tre pazienti hanno mostrato segni di maturazione, con miglioramento le soglie di intervento del tronco cerebrale uditive, sia da nessuna risposta o di risposta solo ai volumi elevati, ad una risposta rilevabile a basso volume. Un bambino nel gruppo neuropatia non uditiva, nato a 29 settimane di gestazione “, che aveva una malattia polmonare cronica, la sepsi e l’esposizione gentamicina, aveva prove di sentire maturazione a 7 mesi di età. Due bambini con neuropatia uditiva avevano evidenza di maturazione udito: uno a 10 mesi, che erano nati a 37 settimane di gestazione ‘e sviluppate kernicterus e CP; l’altra a 7 mesi, che erano nati a 29 settimane di gestazione ‘e sviluppato ipossia, emorragia intraventricolare, sepsi e malattia polmonare cronica.
Attualmente sette dei bambini affetti da neuropatia non uditiva e solo uno del gruppo neuropatia uditiva hanno subito l’impianto cocleare, ad un’età media di 1 anno 7 mesi (range 10mo-2y 8mo). Queste cifre possono cambiare come più dei bambini più piccoli completare le loro valutazioni.
Discussione
Fattori di rischio per ipoacusia sono stati ben documentati. Vohr et al. 8 prospetticamente recensione 4478 bambini ad alto rischio sulle unità di terapia intensiva neonatale e 2701 su unità ben infantile, (2348 neonati senza fattori di rischio e 353 con fattori di rischio). Nella popolazione unità di terapia intensiva neonatale, i quattro fattori di rischio più comuni erano farmaci ototossici (44,4%), molto basso peso alla nascita (17,8%), ventilazione assistita per più di 5 giorni (16,4%) e bassi punteggi di Apgar a 1 o 5 minuti (13,9%). I sei fattori di rischio più comuni nei vivai ben infantili, erano una storia familiare (6,6%), anomalie cranio-facciali (3,4%), bassi punteggi Apgar (2,8%), sindromi o stigmate associati alla perdita di udito (0,5%), i farmaci ototossici ( 0,2%) e infezione congenita (0,1%). I pazienti con neuropatia uditiva comprendono un sottogruppo di pazienti con insufficienza uditiva, coinvolgendo diverse patologie più comunemente associati con i fattori di rischio connessi all’ammissione alla unità di terapia intensiva neonatale.
Anche se i pazienti possono presentarsi con nessuno dei fattori di rischio noti, nella nostra serie di pazienti con neuropatia uditive, i fattori di rischio più significativi sembrano essere iperbilirubinemia, emorragia intracranica, sepsi, e il trattamento gentamicina. Questi sono stati identificati in altre serie clinica di neuropatia uditiva con tassi di iperbilirubinemia di 50-73%, nascita pretermine del 30 al 46%, esposizione al farmaco ototossica di 41 a 80%, storia familiare di perdita di 36 udito al 38%, meccanico ventilazione di 36%, e CP di 9 a 15%. 2,9,10
Non tutti gli studi hanno suscitato questi fattori. Berg et al 11 hanno studiato i pazienti ricoverati in una terapia intensiva pediatrica, guardando sesso, età gestazionale, peso alla nascita, difficoltà respiratoria, iperbilirubinemia, uso di farmaci potenzialmente ototossici, nascite multiple, convulsioni, e la storia della famiglia. Un confronto tra bambini con e senza neuropatia uditiva non ha mostrato una differenza significativa l’esposizione o la presenza di fattori di rischio, oltre a iperbilirubinemia, vancomicina, e furosemide.
Anche se sono stati trovati neonati con neuropatia uditiva ad avere livelli significativamente più elevati di specifici neuroni enolasi (indicatore di danno neuronale) rispetto a quelli senza neuropatia uditiva, nessuna correlazione è stata trovata tra i livelli enolasi neurone-specifica e livelli di bilirubina sierica. 12
In studi istopatologici, perdita di cellule ciliate interne hanno sempre stati trovati con iperbilirubinemia 9 e farmaci ototossici. 13,14 iperbilirubinemia è stato trovato per causare grave degenerazione dei neuroni gangliari a spirale e una scarsità di assoni mielinizzati in un itterico modello di ratto Gunn a fronte di un non itterico modello. 15
In molti casi, i tempi per la diagnosi e il primo utilizzo di un apparecchio acustico sono lunghi perché alcuni dei bambini erano troppo bene o immaturo per essere testato o avere apparecchi montati udito.
A causa delle incertezze che circondano la prognosi di neuropatia uditiva, le principali problematiche nella gestione dei bambini con questa diagnosi sono la fornitura di informazioni e sostegno per la famiglia, ripetuta valutazione audiologica (sia obiettivo [elettrofisiologica] e comportamentale), valutazione della comunicazione del bambino e le decisioni circa la fornitura di amplificazione (in termini di un apparecchio acustico convenzionale o impianto cocleare) e altre gestione medica. Questi requisiti sono complessi e non possono essere forniti da una o due persone. Quindi un team multidisciplinare di professionisti tra cui fisici medici, gli insegnanti dei sordi, audiologi, medici audiovestibular, logopedisti, comunità pediatri dello sviluppo, neurologi pediatrici, otorinolaringoiatri, e la famiglia si dovrebbe intraprendere la diagnosi e il trattamento di questi bambini.
Molti bambini con neuropatia uditiva sono in grado di fare buon uso del loro udito e beneficiare di-audioprotesisti, se vi è sufficiente la perdita dell’udito comportamentale per garantire la fornitura di amplificazione. 2 Tuttavia, in alcuni casi, anche se ci può essere un miglioramento nella rilevazione e la consapevolezza del suono e della parola, il beneficio a lungo termine di apparecchi acustici in termini di comprensione del parlato può essere molto povera e non paragonabile al vantaggio ottenuto in un bambino con una neuropatia non uditiva sordità neurosensoriale di grado simile. 2, 16
Dopo la diagnosi di neuropatia uditiva si è soliti effettuare una prova con gli apparecchi acustici in cui ci sono soglie uditive comportamentale affidabili e cresciuto. Come le soglie di intervento del tronco encefalico uditive elettrofisiologiche potrebbero differire significativamente da soglie comportamentali udienza, la prescrizione apparecchio acustico dovrebbe essere basato sul comportamento delle valutazioni audiologiche. 2 Nel caso in cui la valutazione del comportamento è difficile o dubbia, osservazioni soggettive da parte della famiglia e professionisti del la risposta del bambino a uso di un apparecchio acustico vengono utilizzati.
Sorprendentemente, la letteratura ha mostrato risultati positivi in termini di rilevazione e comunicazione per alcuni bambini con diagnosi di neuropatia uditiva che ricevono un impianto cocleare. 16,18,19 Questo implica integrità funzionale del nervo prossimale ottava (cerebrali reparti) al impianto in questi casi. Tuttavia, i risultati sono variabili. 20,21 preoccupazioni per impiantazione includono il potenziale di maturazione con miglioramento e la patologia essendo più prossimale, rendendo pertanto la stimolazione tramite l’impianto inutile. È, quindi, di solito offrire una prova con amplificazione convenzionali apparecchi acustici prima considerazione di impianto cocleare. La modalità di comunicazione che il bambino, in famiglia dovrebbe essere una scelta informata dalla famiglia se stessi. Tuttavia, gli ingressi visivi come comunicazione totale, firmato o discorso cued sono opzioni efficaci per i bambini con diagnosi di neuropatia uditiva. 22
Conclusione
La nostra comprensione della neuropatia uditiva è in crescita, come si comincia a riconoscere i fattori di rischio e la patologia in questione, più pazienti vengono diagnosticati, e le opzioni di trattamento sono sviluppati. Anche se è un problema raro, può essere sottodiagnosticata a causa della mancanza di familiarità da parte dei professionisti e il modo in cui attualmente ci schermo neonati, anche se lo screening universale con risposta uditivi del tronco encefalico non è una valida opzione.
In questa popolazione, neuropatia uditiva è molto più comune di quanto finora previsto, e gli infanti in generale hanno avuto più grave malattia neonatale rispetto a quelli con i non-uditivi menomazioni dell’udito neuropatia. Tuttavia, vi è una reale possibilità di miglioramento nel tempo come il bambino matura, almeno in alcuni casi, e in seguito sistematico è necessario per quantificare questo.
References
- 1
, , , . Absent auditory brain stem response: peripheral hearing loss or brain stem dysfunction? Laryngoscope 1984 ; 94 : 400 – 06 .
, , , et al. Clinical findings for a group of infants and young children with auditory neuropathy . Ear Hear 1999 ; 20 : 238 – 52 .
, . The effects of hypoxia on sensory cells of the cochlea in chinchilla . Scanning Microsc 1987 ; 1 : 1175 – 83 .
, , , , . Induction of selective inner hair cell damage by carboplatin . Scanning Microsc 1994 ; 8 : 97 – 106 .
, , . Pathophysiology of auditory neuropathy . In: SiningerY , StarrA , editors. Auditory neuropathy: a new perspective on hearing disorders . Canada: Singular, Thomson Learning Inc, 2001 : 67 – 82 .
- 6
, , , , . Screening and follow up assessment in three cases of auditory neuropathy . Arch Dis Child 2003 ; 88 : 25 – 26 .
. NHS newborn hearing screening programme: auditory neuropathy/auditory dys-synchrony – timeline of investigation and management . http://hearing.screening.nhs.uk/getdata.php?id=147 (accessed 11 November 2008).
- 8
, , , et al. Identification of neonatal hearing impairment: characteristics of infants in the neonatal intensive care unit and well-baby nursery . Ear Hear 2000 ; 21 : 373 – 82 .
, , , et al. Selective inner hair cell loss in premature infants and cochlea pathological patterns from neonatal intensive care unit autopsies . Arch Otolaryngol Head Neck Surg 2001 ; 127 : 629 – 36 .
- 10
, , , , . Clinical and audiological features in auditory neuropathy . Arch Otolaryngol Head Neck Surg 2002 ; 128 : 1026 – 30 .
, , , , . Newborn hearing screening in the NICU: profile of failed auditory brainstem response/passed otoacoustic emission . Pediatrics 2005 ; 116 : 933 – 38 .
, , , , , . Auditory neuropathy in hyperbilirubinemia: is there a correlation between serum bilirubin, neuron-specific enolase levels and auditory neuropathy? In J Audiol 2004 ; 43 : 516 – 22 .
. An animal model of auditory neuropathy . Ear Hear 1998 ; 19 : 355 – 61 .
- 14
, , , , . Auditory deprivation of the central auditory system resulting from selective inner hair cell loss: animal model of auditory neuropathy . Scand Audiol Suppl 1999 ; 51 : 1 – 12 .
, , . The jaundiced Gunn rat model of auditory neuropathy/dyssynchrony . Laryngoscope 2005 ; 115 : 2167 – 73 .
Direct Link:
, , , . Speech perception and cortical event related potentials in children with auditory neuropathy . Ear Hear 2002 ; 23 : 239 – 53 .
, , , , . Cochlear implants in five cases of auditory neuropathy: prospective findings and progress . Laryngoscope 2001 ; 111 : 555 – 62 .
Direct Link:
, , , , , . Auditory neuropathy/dys-synchrony: after the diagnosis, then what? Semin Hear 2002 ; 23 : 209 – 14 .
- 19
, , . Cochlear implantation of auditory neuropathy . J Am Acad Audiol 2000 ; 11 : 309 – 15 .
, , , . Cochlear implantation in auditory neuropathy . Laryngoscope 1999 ; 109 : 181 – 85 .
Direct Link:
, , , , , . Outcome of cochlear implantation in pediatric auditory neuropathy . Otol Neurotol 2002 ; 23 : 328 – 32 .
, , , et al. Assessment and management of auditory neuropathy/auditory dys-synchrony. A recommended protocol . Version 1.1 May 2008. http://hearing.screening.nhs.uk/getdata.php?id=9641(accessed 11 November 2008).