GENERALITA’ E CLASSIFICAZIONI

Le sordità genetiche sono causate da un’alterazione del materiale genetico. Il materiale genetico, DNA (acido desossiribonucleico), è contenuto in quasi tutte le cellule del corpo umano. Le lunghe catene di DNA possono essere suddivise in sezioni codificanti chiamate geni. Ogni individuo eredita due copie di ogni gene, una copia da ogni genitore. I geni controllano la produzione e la funzione di proteine, che a loro volta determinano la struttura e la regolazione del corpo. Sono costituiti da una specifica sequenza di 4 unità chimiche (“basi”) adenina (A), guanina (G), citosina (C), timina (1), sono abbastanza simili da individuo a individuo, L’uomo possiede circa 30 000 geni (molto meno di quello che si riteneva alcuni anni fa), una frazione dei quali si ritiene implicato nella “costruzione” e nel funzionamento (omeostasi) dell’orecchio interno.
Come si è detto nel capitolo della sordità infantile, si stima che circa metà delle sordità presenti alla nascita siano dovute a fattori genetici. Tali forme possono essere di gravità diversa. Tuttavia una sordità genetica può anche manifestars i a qualsiasi età della vita, ed essere variamente progressiva. Per queste forme, definite ad esordio tardivo, non esistono stime di prevalenza, anche se si sospetta che siano responsabili di una proporzione considerevole delle ipoacusie neurosensoriali.
Le sordità genetiche possono essere classificate secondo vari criteri, Il primo di essi è essenzialmente clinico e contempla due grandi categorie: forme non sindromiche e forme sindromiche.
· forme non sindromiche in cui la perdita dell’udito non è accompagnata da altri sintomi e comprendono circa il 70% dei casi. Due terzi di queste forme presenta una trasmissione di tipo autosomico recessivo, un terzo è legato ad una trasmissione di tipo autosomico dominante, mentre solo l’1-2% è riconducibile ad una trasmissione associata al cromosoma X. Una percentuale ancora da definire è dovuta a mutazioni del DNA mitocondriale.
· forme sindromiche in cui la perdita dell’udito di associa ad altre manifestazioni cliniche e comprendono il restante 30% dei casi. Tra le forme sindromiche più comuni vi sono la sindrome di Pendred (sordità e gozzo) e la sindrome di Usher (sordità e cecità progressiva).
Nella maggior parte dei casi (circa 70-80%) di sordità genetica non sindromica a trasmissione autosomica recessiva è coinvolto il gene della connessina 26 (Cx26, noto anche come GJB2, gap-junction protein beta 2). Le alterazioni a carico di questo gene sono numerose, ma la mutazione più frequente (50-80% dei casi) è nota come 30/35delG. La frequenza dei portatori sani (eterozigoti asintomatici) in Italia è di 1 su 35 individui. Due genitori portatori sani (asintomatici) avranno una probabilità del 25% di avere figli affetti da sordità genetica non sindromica. Dalla stessa unione i figli avranno una probabilità del 50% di nascere portatori sani, come i genitori. Il gene Cx26 codifica per la connessina 26, una proteina di giunzione che forma dei canali per mettere in comunicazione fra loro le cellule cocleari e consentire il passaggio di mediatori chimici, fra i quali lo ione potassio, fondamentali per la funzionalità dell’orecchio. Un’altra mutazione del gene GJB2, meno frequente della 30/35delG, è la 167delT. Un altro gene associato a sordità non sindromica è Cx30 o GJB6 (gap-junction protein beta 6) che codifica per la connessina 30. Studi pubblicati hanno evidenziato la presenza di una mutazione (D13S1830)che consiste in una delezione all’interno del gene. La delezione di GJB6 sia in omozigosi (entrambe le copie del gene sono mutate) che in associazione con una mutazione del gene Cx26 (eterozigosi composta) causano perdita dell’udito.

Ad oggi sono state identificate più di 400 tipi diversi di sindromi in cui è coinvolta la funzione uditiva. Si ricorda che una sindrome è un gruppo di sintomi o caratteri patologici che riconoscono lo stesso fattore causale.
Questi altri disordini associati alla sordità interessano:
 1. struttura anatomica cranio-facciale
1. struttura anatomica cranio-facciale
2. apparato oculo-visivo
3. apparato muscolo-scheletrico
4. apparato tegumentario (cute ed annessi)
5. apparato renale
6. sistema nervoso centrale
7. sistema metabolico-endocrino
Una seconda classificazione è basata sulla modalità con cui si eredita la sordità (Fig 1)
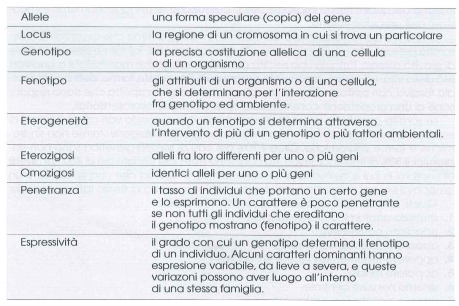
Nella trattazione della trasmissione ereditaria dei caratteri si ricorre ad una peculiare terminologia La tabella IV elenca le principali definizioni.
Trasmissione autosomica recessiva
TRASMISSIONE AUTOSOMICA RECESSIVA
Le sordità genetiche si possono trasmettere secondo diverse modalità.
Autosomica recessiva (AR)
In questa modalità di trasmissione i due alleli del gene devono essere muti perché il soggetto sia sordo. I genitori portatori di una copia anomala (allele) del gene in causa (portatori eterozigoti) sono normoudenti e, statisticamente, un quarto dei bambini (maschio o femmina) saranno sordi, portatori di mutazioni sui due alleli del gene (omozigoti). La modalità di trasmissione autosomica recessiva è favorita dalla consanguineità. Si stima che circa tre quarti delle sordità non sindromiche si trasmettano in modalità AR e che tale modalità sia la seconda in frequenza nelle sordità sindromiche.

Figura 1A: trasmissione ereditaria di una malattia autosomica recessiva. Il sesso non incide, in quanto la mutazione genetica risiede su un cromosoma non sessuale (o autosomico).
Ipoacusia Autosomica recessiva (AR) (76%)
E’ la modalità più frequente. infatti si stima che circa tre quarti delle ipoacusie non sindromiche si trasmettano in modalità AR. Quando una malattia si trasmette in ipoacusia a trasmissione autosomica recessiva della moda, due copie del gene anomalo causano la malattia. Gli individui che ereditano un solo gene anomalo e un gene normale sono indicati come vettori e non sono colpiti dalla malattia. I genitori portatori di un solo allele anomalo del gene in causa (portatori eterozigoti) Sono normoudenti e spesso sono inconsapevoli del loro stato di portatore fino a quando non hanno un figlio affetto. Gli individui affetti da una malattia autosomica recessiva di solito sono il risultato di accoppiamenti tra due vettori. La Fig.2 illustra il potenziale risultati di tale accoppiamento, in cui il 25% della prole sono affetti da disturbo in questione, il 50% sono portatori sani, e il 25% sono liberi della malattia e con il gene anormale. La Consanguineità , o la presenza di un comune antenato tra la coppia, aumenta il rischio di una malattia autosomica recessiva. La Sindrome di Usher mostra un modello di ipoacusia autosomica recessiva. La prevalenza della sindrome di Usher è di 3 a 4 soggetti affetti si 100.000 nati (Ahmed, Riazuddin, Riazuddin, & Wilcox, 2003), con tassi di prevalenza più elevati in alcune popolazioni, tra cui la popolazione Acadian in Louisiana (Ouyang et al., 2003) e la popolazione ebrea Ashkenazi (Guha et al., 2012). Il tipo più grave di sindrome di Usher è caratterizzata da una sordità profonda neurosensoriale congenita e da progressiva perdita della vista (Liu et al., 2013). Garantire una diagnosi precoce della sindrome di Usher può aiutare i pazienti a prepararsi per il loro futuro. La sindrome di Usher può rappresentare la metà di tutti i casi di concomitante sordità / cecità (Yan e Liu, 2010).

Fig.2 ereditarietà autosomica recessiva

Sordità sindromiche EMC
“Le sordità sindromiche rappresentano solo una piccola percentuale di sordità del bambino (10 – 15% circa) e una porzione poco conosciuta, probabilmente inferiore, di sordità dell’adulto. Sono state descritte diverse centinaia di sindromi con sordità (vedi [8]), e a tutt’oggi è stato identificato più di un centinaio di geni. È comunque importante conoscere e ricercare le principali sindromi, poiché la gestione e la valutazione eziologica saranno differenti da una sordità non sindromica. In ragione del grande numero di sindromi rare con sordità qualsiasi patologia malformativa nel bambino deve far effettuare un esame uditivo sistematico. Per le sordità sindromiche inoltre che comprendono una lesione malformativa craniofaciale, la sordità è molto spesso aggravata da un’otite cronica e s’impone un monitoraggio otologico regolare.
Abbiamo elencato nella Tabella 4 le sette sordità sindromiche che ci sembrano più importanti e che devono essere conosciute dai diversi specialisti che assumono in carico le sordità, a motivo della loro frequenza e/o della loro gravità potenziale. La Tabella 5 elenca in modo più completo le sindromi no eccezionali i cui geni sono stati identificati (per una rassegna esauriente dei geni clonati della sordità sindromica, vedi [9]). Descriveremo qui le sette sindromi della Tabella 1.
Tre sindromi autosomiche recessive
Tre sindromi autosomiche recessive devono essere sistematicamente ricercate: le sindromi di Pendred e di Usher, entrambe frequenti, e la rara sindrome di Jerwell e Lange-Nielsen. Tutte queste sindromi hanno la particolarità di presentarsi a lungo come una sordità isolata, e solo una valutazione sistematica può permettere di individuarle precocemente.
Sindrome di Pendred
Questa sindrome è stata descritta da oltre 100 anni. È riconosciuta dall’associazione di una sordità di origine cocleare, il più delle evolutiva, prelinguale o postlinguale precoce, con un disturbo del metabolismo dello iodio che si manifesta con un gozzo tiroideo. Essa si trasmette un modo autosomico recessivo. Il gene in causa, PDS (attualmente chiamato SLC26A4), è implicato sia nella sindrome di Pendred [10] che in una forma di sordità che rimane isolata: DFNB4. [11, 12] Il confine tra la sindrome di Pendred e la forma di sordità isolata DNFB4 è a volte difficile da definire poiché, all’interno di famiglie affette da Pendred, uno o più individui possono non sviluppare la lesione tiroidea. [13] Noi descriveremo la forma DFNB4 nel paragrafo «Sordità non sindromiche».
La prevalenza della sindrome di Pendred è stimata a 7-10 cas/ 100.000 nascite. La sordità ha la particolarità, quando non è molto profonda fin dall’inizio, di evolvere per gradi di peggioramento improvviso, seguiti da un recupero in genere parziale. Queste fluttuazioni sono estremamente invalidanti e angosciose per il bambino.
L’età di comparsa del gozzo tiroideo è variabile, il più delle volte, nel corso del secondo decennio di vita (da 6 a 37 anni con età media 14,9 anni nella recente casistica di Blons sul territorio francese) e in questa stessa casistica era associato un ipotiroidismo nel 77% dei casi. [14] La sindrome di Pendred si presenta quindi come una sordità isolata per molti anni.
La TC delle rocche mette sempre in evidenza anomalie morfologiche dell’orecchio interno (dilatazione dell’acquedotto del vestibolo (Figura 2 ), e a volte coclea incompleta e dilatata di tipo «Mondini») in modo quasi costante, ma queste anomalie possono essere monolaterali. [15] Le anomalie di organificazione dello iodio possono essere messe in evidenza con la scintigrafia tiroidea con test al perclorato: l’incorporazione degli ioduri alla molecola di tireoglobulina avviene in modo anormale e la somministrazione di perclorato, anione inorganico, induce un rilascio di ioduri non organificati. La misura della quantità di iodio radioattivo prima e dopo somministrazione di perclorato mostra una diminuzione superiore al 10%. Questo test può permettere di individuare una sindrome di Pendred prima della comparsa di gozzo nei pazienti con malformazione dell’orecchio interno. Tuttavia questo tipo di test non è né sensibile (normale in certi soggetti in alcune forme familiari) né specifica (positivo in particolare nella tiroidite di Hashimoto e nei pazienti con mutazioni dei geni della tiroide perossidasi o della tiroglobulina). Ha inoltre l’inconveniente di sottoporre il paziente a irradiazione.
Il gene responsabile della sindrome di Pendred è stato localizzato nel 1997 nella regione 7q31 [16,17] ed è stato clonato da Everett e coll.. [10] Il gene PDS codifica per la pendrina formata da 780 aminoacidi. A tutt’oggi sono state identificate più di cinquanta mutazioni nel gene PDS. Queste mutazioni sono ripartite nei 21 esoni del gene. Quattro di esse sono particolarmente frequenti, presenti sul 74% dei cromosomi trasferiti. [14, 18] Mutazioni della PD S sono evidenziate nella grande maggioranza dei pazienti che presentano una sindrome di Pendred in Francia ed è sempre presente una dilatazione dell’acquedotto del vestibolo nei soggetti in cui sono identificate le due mutazioni. [14]
Sindrome di Usher
La sindrome di Usher associa alla sordità una retinite pigmentosa. Esistono molteplici forme di sindromi di Usher, ma i tre quarti sono Usher di tipo I con sordità congenita profonda, areflessia vestibolare bilaterale responsabile di un ritardo della deambulazione (deambulazione dopo 18 mesi) e retinite che si sviluppa durante l’infanzia. I primi segni visivi sono dei disturbi della visione in penombra, spesso verso l’inizio del secondo decennio di vita, ma l’esame sistematico del fondo dell’occhio può consentire la diagnosi molto prima di questa età, fin dai 3-4 anni. L’esame più precoce è l’elettroretinogramma, patologico prima dei primi segni nel fondo dell’occhio. La sindrome di Usher di tipo I è un’indicazione all’impianto cocleare precoce per ottenere una comprensione del linguaggio senza lettura labiale in questi bambini che, in età adulta, avranno una rilevante compromissione visiva. Porre la diagnosi attraverso l’esame del fondo dell’occhio a 4 anni è quindi già una situazione tardiva. In linea di principio l’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi, e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale.
CONDOTTA DA TENERE
L’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale.
Nella sindrome di Usher tipo II la sordità è in media grave, non progressiva, predominante sulle frequenze acute, la retinite un poco più tardiva e i segni vestibolari assenti. Nella Usher tipo III la sordità è progressiva, i segni vestibolari e l’età di inizio della retinite sono variabili (per una rassegna, vedi [8]).
A tutt’oggi sono stati localizzati 11 geni e otto di questi geni sono stati identificati, cinque per la sindrome di Usher tipo I, due per il tipo II e uno per il tipo III (Tabella 4). MYO7A e CDH23rappresentano rispettivamente il 30 e 29% dei casi di Usher tipo I e USH2A il 40% dei tipi II. [19] La diagnosi molecolare non è eseguita di routine e la diagnosi è essenzialmente clinica.
Sindrome di Jerwell e Lange-Nielsen
È rara (1/100.000), ma di diagnosi facile tramite un elettrocardiogramma (ECG) sistematico in caso di sordità grave o profonda congenita. [20] L’ECG mostra un allungamento dello spazio QT che traduce un disturbo della conduzione cardiaca, fonte di malessere o di morte improvvisa che possono manifestarsi senza circostanze scatenanti o in seguito a uno stress. La prevenzione è possibile mediante un trattamento medico. Due geni sono stati identificati, KCNQ1 e KCNE1, [20, 21, 22, 23] che codificano per alcuni canali del potassio localizzati nella stria vascolare (Tabella 4). I genitori portatori eterozigoti della mutazione possono presentare dei malesseri (donde l’importanza dell’anamnesi familiare) e un’ipoacusia.”
BIBLIOGRAFIA
|
[8] |
Gorlin R.J., Toriello H.V., Cohen M.M. Hereditary hearing loss and its syndromes New York: Oxford University Press (1995). |
||
|
[9] |
Marlin S. Surdités génétiques Génétique médicale Paris: Masson (2004). 281-299 |
||
|
[10] |
Everett L.A., Glaser B., Beck J.C., Idol J.R., Buchs A., Heyman M., e al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat. Genet. 1997 ; 17 : 411-422 [cross-ref] |
||
|
[11] |
Li X.C., Everett L.A., Lalwani A.K., Desmukh D., Friedman R.B., Green E.D., e al. A mutation in PDS causes non-syndromic recessive deafness Nat. Genet. 1998 ; 18 : 215-217 [cross-ref] |
||
|
[12] |
Usami S., Abe S., Weston M.D., Shinkawa H., Van Camp G., Kimberling W.J. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations Hum. Genet. 1999 ; 104 : 188-192 [cross-ref] |
||
|
[13] |
Van Hauwe P., Everett L.A., Coucke P., Scott D.A., Kraft M.L., Ris-Stalpers C., e al. Two frequent missense mutations in Pendred syndrome Hum. Mol. Genet. 1998 ; 7 : 1099-1104 [cross-ref] |
||
|
[14] |
Blons H., Feldmann D., Duval V., Messaz O., Denoyelle F., Loundon N., e al. Screening of SLC26A4 (PDS) gene in Pendred’s syndrome: a large spectrum of mutations in France and phenotypic heterogeneity Clin. Genet. 2004 ; 66 : 333-340 [cross-ref] |
||
|
[15] |
Phelps P.D., Coffey R.A., Trembath R., Luxon L., Grossman A., Britton K.E., e al. Radiological malformations of the ear in Pendred syndrome Clin. Radiol. 1998 ; 53 : 268-273 [cross-ref] |
||
|
[16] |
Coyle B., Coffey R., Armour J.A., Gausden E., Hochberg Z., Grossman A., e al. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4 Nat. Genet. 1996 ; 12 : 421-423 [cross-ref] |
||
|
[17] |
Sheffield V.C., Kraiem Z., Beck J.C., Nishimura D., Stone E.M., Salameh M., e al. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification Nat. Genet. 1996 ; 12 : 424-426 [cross-ref] |
||
|
[18] |
Coyle B., Reardon W., Herbrick J.A., Tsui L.C., Gausden E., Lee J., e al. Molecular analysis of the PDS gene in Pendred syndrome (sensorineural hearing loss and goitre) Hum. Mol. Genet. 1998 ; 7 : 1105-1112 [cross-ref] |
||
|
[19] |
Ouyang X. Mutational analysis of the six cloned Usher genes using a sequential screening strategy Florida: Communication, 26th ARO meeting (2000). |
||
|
[20] |
Splawski I., Timothy K.W., Vincent G.M., Atkinson D.L., Keating M.T. Molecular basis of the long-QT syndrome associated with deafness N. Engl. J. Med. 1997 ; 336 : 1562-1567 [cross-ref] |
||
|
[21] |
Neyroud N., Richard P., Vignier N., Donger C., Denjoy I., Demay L., e al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome Circ. Res. 1999 ; 84 : 290-297 |
||
|
[22] |
Wang Z., Li H., Moss A.J., Robinson J., Zareba W., Knilans T., e al. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome Mol. Genet. Metab. 2002 ; 75 : 308-316 [cross-ref] |
||
|
[23] |
Chen Q., Zhang D., Gingell R.L., Moss A.J., Napolitano C., Priori S.G., e al. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome Circulation 1999 ; 99 : 1344-1347 |
||
APPROFONDIMENTO
Pendrina e Sindrome di Pendred La sindrome Gozzo-sordità Sordità con gozzo
– trasmissione autosomica recessiva
– sordità neurosensoriale progressiva, esordio prelinguale-precoce
– alcuni individui con gozzo, ma normale funzionale tiroidea
– Sindrome dell’acquedotto vestibolare allargato (EVAS) e displasia cocleare (Mondini)
|
Sindrome di Pendred
|
Modalità di trasmissione |
Tipo di deficit uditivo |
Anomalie associate |
|
1-10% delle sordità |
A. R. gene SLC26A4(cr 7), gene FOXI1 (cr.5) |
Sordità neurosensoriale, generalmente congenita o a insorgenza nei primi anni di vita e progressiva |
Disfunzione tiroidea e gozzo Associazione con malformazioni dell’orecchio interno, quali dilatazione acquedotto vestibolare e sacco endolinfatico, malformazione di Mondini |
Che cosa è la sindrome di Pendred?
Come la sindrome di Pendred ha effetto su altre parti del corpo?
Che cosa provoca la sindrome di Pendred?
Come viene diagnosticata la sindrome di Pendred?
Come può essere trattata la sindrome di Pendred?
Che cosa è la sindrome di Pendred?
E’ causata da mutazioni (documentati 50 tipi diversi) del gene PDS che codifìca per una proteina “pendrina” responsabile della riduzione in forma organica dello iodio Lo stesso gene, come si è visto sopra, è anche implicato in una forma di sordità isolata, recessiva, DFNB4, è il secondo tipo più comune di perdita di udito autosomica recessiva sindromica, LA sordità all’inizio può non essere grave, ma poi può progredire con fluttuazioni caratteristiche, aggravamenti seguiti da recuperi parziali. Questi fenomeni sono in relazione ai difetti strutturali del labirinto osseo (EVA, coclea incompleta e dilatata) che impediscono l’omeostasi endo-perilinfatica La sindrome di Pendred è una malattia genetica che provoca la perdita precoce dell’udito nei bambini. La malattia può anche interessare la ghiandola tiroide e, a volte crea problemi di equilibrio. La sindrome prende il nome da Vaughan Pendred, il medico che per primo descrisse persone con il disordine.
I bambini che sono nati con la sindrome di Pendred possono cominciare a perdere l’udito alla nascita o dai tre anni di età. Di solito, il loro udito peggiora nel tempo. La perdita di udito spesso avviene all’improvviso, anche se alcuni individui in seguito recuperano un po’ di udito. Alla fine, alcuni bambini con la sindrome di Pendred diventano totalmente sordi.
Quasi tutti i bambini con sindrome di Pendred hanno perdita di udito bilaterale , il che significa perdita di udito in entrambe le orecchie, anche se un orecchio può avere una maggiore perdita di udito rispetto all’altro.
La perdita dell’udito infantile ha molte cause. I ricercatori ritengono che negli Stati Uniti 50 a 60 percento dei casi sono dovuti a cause genetiche, e dal 40 al 50 percento dei casi derivino da cause ambientali. Gli operatori sanitari utilizzano diversi indizi, come ad esempio quando inizia la perdita di udito e se ci sono differenze anatomiche nelle orecchie, per aiutare a determinare se un bambino ha la sindrome di Pendred o qualche altro tipo di sordità progressiva.
 Questa sindrome è stata descritta da oltre 100 anni. Fu descritta nel 1896 dal Dr. Vaughan Pendred, medico inglese, il quale, per la prima volta, segnalò il caso di una famiglia irlandese che viveva aDurhamnel 1896 Pendred V. ,Pearce JM 2007, con due sorelle (di dieci figli) affette da sordomutismo congenito e gozzo tiroideo, non correlabili in alcun modo a fattori acquisiti o ambientali.
Questa sindrome è stata descritta da oltre 100 anni. Fu descritta nel 1896 dal Dr. Vaughan Pendred, medico inglese, il quale, per la prima volta, segnalò il caso di una famiglia irlandese che viveva aDurhamnel 1896 Pendred V. ,Pearce JM 2007, con due sorelle (di dieci figli) affette da sordomutismo congenito e gozzo tiroideo, non correlabili in alcun modo a fattori acquisiti o ambientali.
Nonostante il Dr. Pendred intuì l’ereditarietà della sindrome, solo nel 1956 fu dimostrata la modalità di trasmissione autosomica recessiva del fenotipo.
Due anni più tardi, si attribuì il gozzo tiroideo a difetti di organificazione dello iodio a livello tiroideo con conseguente deficit di sintesi di tiroxina.
Nel 1967 e nel 1978 vennero descritte le tipiche malformazioni dell’orecchio interno della sindrome di Pemdred ossia la malformazione tipo Mondini (con appiattimento della coclea e sviluppo del solo giro basale) e la dilatazione dell’acquedotto vestibolare, quest’ultima di gran lunga più comune e frequente.
Dal punto di vista epidemiologico, la sindrome di Pendred ha un’incidenza di circa 9 affetti ogni 100.000 nuovi nati.
La sindrome, non curabile, non compromette la prognosi quoad vitam del paziente, né tanto meno le sue capacità riproduttive. Si suppone, ad oggi, che dall’1 all’8% delle ipoacusie congenite sia attribuibile alla Sindrome di Pendred.
È riconosciuta dall’associazione di una sordità di origine cocleare, il più delle evolutiva, prelinguale o postlinguale precoce, con un disturbo del metabolismo dello iodio che si manifesta con un gozzo eutiroideo. Il gozzo si sviluppa nella pubertà precoce o nell’età adulta. Essa si trasmette un modo autosomico recessivo. durante la valutazione radiografica si trovano comunemente la displasia di Mondini e la dilatazione dell’ acquedotto vestibolare E’ causata da mutazioni (documentati 50 tipi diversi) del gene PDS che codifìca per una proteina “pendrina” responsabile della riduzione in forma organica dello iodio Lo stesso gene, come si è visto sopra, è anche implicato in una forma di sordità isolata, recessiva, DFNB4,
Quanto è diffusa la sindrome di Pendred?
Il gene SLC26A4, che causa la sindrome di Pendred, rappresenta circa il 5-10 per cento di perdita dell’udito ereditaria. Dal punto di vista epidemiologico, la sindrome di Pendred ha un’incidenza di circa 9 affetti ogni 100.000 nuovi nati.
Man mano che i ricercatori acquisiranno maggiori conoscenze sulla sindrome e sulle sue caratteristiche, si spera di migliorare la capacità dei medici di rilevare e diagnosticare la malattia.
La sindrome di Pendred viene ereditata in maniera autosomica recessiva, il che significa che è necessario ereditare il gene anomalo da ciascun genitore affinché la malattia si possa manifestare. Ciò significa anche che un fratello di un paziente con la sindrome di Pendred ha una probabilità del 25% di avere anch’esso la condizione.

La sordità all’inizio può non essere grave, ma poi può progredire con fluttuazioni caratteristiche, aggravamenti seguiti da recuperi parziali. Questi fenomeni sono in relazione ai difetti strutturali del labirinto osseo (EVA, coclea incompleta e dilatata) che impediscono l’omeostasi endo-perilinfatica
Che cosa provoca la sindrome di Pendred?
Il gene in causa, PDS (attualmente chiamato SLC26A4) è localizzato sul braccio lungo del cromosoma 7 (7q31)( Sheffield VC., ed Al19896; Coyle B., ed Al.,1996), è implicato sia nella sindrome di Pendred (Everett L.A.),che in una forma di sordità che rimane isolata: DFNB4(Li X.C., Usami S.). Il confine tra la sindrome di Pendred e la forma di sordità isolata DNFB4 è a volte difficile da definire poiché, all’interno di famiglie affette da Pendred, uno o più individui possono non sviluppare la lesione tiroidea(Van Hauwe P.). Mutazioni nello stesso gene causano anche la sindrome dell’acquedotto vestibolare allargato (EVAS), un’altra causa della sordità congenita; specifiche mutazioni hanno maggiori probabilità di causare l’EVAS, mentre altre sono più collegate con la sindrome di Pendred( Azaiez H, ed Al.,2007)
La prevalenza della sindrome di Pendred è stimata a 7-10 cas/ 100.000 nascite. La sordità ha la particolarità, quando non è molto profonda fin dall’inizio, di evolvere per gradi di peggioramento improvviso, seguiti da un recupero in genere parziale. Queste fluttuazioni sono estremamente invalidanti e angosciose per il bambino.
L’età di comparsa del gozzo tiroideo è variabile, il più delle volte, nel corso del secondo decennio di vita (da 6 a 37 anni con età media 14,9 anni nella recente casistica di Blons sul territorio francese) e in questa stessa casistica era associato un ipotiroidismo nel 77% dei casi(Blons H.). La sindrome di Pendred si presenta quindi come una sordità isolata per molti anni.
Segni e sintomi
La perdita dell’udito è spesso, anche se non sempre, presente sin dalla nascita e l’acquisizione del linguaggio può essere un problema significativo se la sordità è grave in età infantile. La perdita dell’udito peggiora in genere nel corso degli anni e la progressione può essere graduale. In alcuni casi lo sviluppo del linguaggio peggiora dopo un trauma cranico, dimostrando che l’orecchio interno è sensibile ai traumi; questa è una conseguenza degli acquedotti vestibolari allargati tipici di questa sindrome(Reardon W, et Al 1997) .La funzione vestibolare varia nella sindrome di Pendred e la presenza di vertigini possono essere una caratteristica di un trauma cranico minore. Il gozzo è presente nel 75% dei casi( Reardon W, et Al 1997).
Come viene diagnosticata la sindrome di Pendred?
Un otorinolaringoiatra (un medico specializzato in malattie di orecchio, naso, gola, testa e collo) o una clinica genetista prenderà in considerazione la perdita, strutture dell’orecchio interno, e, talvolta, la tiroide udito nella diagnosi di sindrome di Pendred.
Lo specialista valuterà la tempistica, l’importo, e il tipo di perdita dell’udito. farà le seguenti domande quali:
-
- “Quando è iniziata la perdita uditiva ?”
-
- “E’ peggiorata nel tempo?”,
- “E’ successo all’improvviso o in stadi?”.
La perdita uditiva precoce è una delle caratteristiche più comuni della sindrome Pendred; tuttavia, solo questo sintomo non significa che un bambino ha la condizione.
Lo specialista utilizzerà interni tecniche di imaging per le orecchie come la risonanza magnetica (MRI) o la tomografia computerizzata (TAC) per cercare due caratteristiche della sindrome di Pendred. Una caratteristica potrebbe essere una coclea con troppo pochi giri. La coclea è la parte a forma di spirale dell’orecchio interno che converte il suono in segnali elettrici che vengono inviati al cervello. Una coclea sana ha due giri e mezzo, ma la coclea di una persona con sindrome di Pendred può avere solo uno giro e mezzo. Non tutte le sindrome di Pendred, tuttavia, hanno una coclea anormale.
L’orecchio interno

NIH Medical Arts
Una seconda caratteristica della sindrome di Pendred è un acquedotto vestibolare allargato (vedi figura). L’acquedotto vestibolare è un canale osseo che va dal vestibolo (parte dell’orecchio interno tra coclea e canali semicircolari) all’interno del cranio. All’interno l’acquedotto vestibolare è un tubo pieno di liquido chiamato il dotto endolinfatico, che termina con il sacco endolinfatico a forma di palloncino. Il dotto endolinfatico e sac di solito sono anche ingrandite.
Gli esperti non è consigliabile testare i livelli di ormone tiroideo nei bambini con sindrome di Pendred, poiché i livelli sono di solito normali. Alcuni bambini potrebbero essere dato un “test di washout perclorato”, un test che determina se la tiroide sta funzionando correttamente. Anche se questo test è probabilmente il miglior test per determinare la funzione tiroidea nella sindrome Pendred, non è usato spesso ed è stato ampiamente sostituito da analisi genetiche. Le persone che hanno sviluppato un gozzo possono essere riferite ad un endocrinologo, un medico specializzato in disturbi ghiandolari, per determinare se il gozzo è dovuto alla sindrome di Pendred o ad altra causa. Gozzo è una caratteristica comune della sindrome di Pendred, ma molte persone che sviluppano un gozzo non hanno la sindrome di Pendred. Al contrario, molte persone che hanno la sindrome di Pendred non sviluppano mai un gozzo.
La diagnosi in età prenatale è effettuabile mediante un esame sul DNA di cellule fetali o mediante l’analisi dei villi coriali.
Successivamente con:
Valutazione audiologica con esami (OAEs, ABR, Impedenzometria, COR, ASSR, audiometria tonale e vocale, quando possibile);
TAC e/o RMN dell’orecchio;
Studio della funzionalità tiroidea ed esame ecografico ghiandolare;
Studio della funzionalità renale ed esame ecografico app urinario (anche se raramente si hanno anomalie);
Test al perclorato di K (Pertiroid); Indagini genetiche (sequenziamento completo, incluse le regioni non tradotte del gene SLC26A4).
Dal punto di vista terapeutico non esistono trattamenti specifici per questo tipo di malattia. Non è possibile infatti, ad oggi, guarire dalle mutazioni del gene SLC26A4, dall’EVA o dalla Sindrome di Pendred, anche se la ricerca sta compiendo passi da gigante nell’ambito della terapia genica e delle cellule staminali.
È possibile però contrastare in maniera efficace e tempestiva i sintomi, tanto da permettere al paziente una vita del tutto normale. Per quanto riguarda l’ipoacusia, ovviamente, il trattamento è rappresentato dalla riabilitazione con protesi acustica o con impianto cocleare. Possono essere d’ausilio anche supporti del linguaggio Per quanto riguarda i disordini della tiroide, qualora dovessero essere presenti, previa valutazione specialistica dell’endocrinologo, è solitamente sufficiente una terapia farmacologica cronica sostitutiva con levo-tiroxina nei casi di gozzo ipotiroideo.
La TC delle rocche mette sempre in evidenza anomalie morfologiche dell’orecchio interno (dilatazione dell’acquedotto del vestibolo (Fig. 2 ), e a volte coclea incompleta e dilatata di tipo «Mondini») in modo quasi costante, ma queste anomalie possono essere monolaterali(Phelps P.D.).
|
|
|
|
La coclea ha normalmente due giri e mezzo, ma nella sindrome di Pendred, ci può essere 1 e mezzo giro solo, a causa della dilatazione abnorme di sacco endolinfatico. |
|
|
|
|
|
Fig. 2 : Dilatazione dell’acquedotto del vestibolo (freccia) su una TC delle rocche in sezione assiale. |
|



Le anomalie di organificazione dello iodio possono essere messe in evidenza con la scintigrafia tiroidea con test al perclorato: l’incorporazione degli ioduri alla molecola di tireoglobulina avviene in modo anormale e la somministrazione di perclorato, anione inorganico, induce un rilascio di ioduri non organificati. La misura della quantità di iodio radioattivo prima e dopo somministrazione di perclorato mostra una diminuzione superiore al 10%. Questo test può permettere di individuare una sindrome di Pendred prima della comparsa di gozzo nei pazienti con malformazione dell’orecchio interno. Tuttavia questo tipo di test non è né sensibile (normale in certi soggetti in alcune forme familiari) né specifica (positivo in particolare nella tiroidite di Hashimoto e nei pazienti con mutazioni dei geni della tiroide perossidasi o della tiroglobulina). Ha inoltre l’inconveniente di sottoporre il paziente a irradiazione.
Il gene responsabile della sindrome di Pendred è stato localizzato nel 1997 nella regione 7q31( Coyle B., Sheffield V.C.), ed è stato clonato da Everett e coll. (Everett L.A.).Il gene PDS codifica per la pendrina formata da 780 aminoacidi. A tutt’oggi sono state identificate più di cinquanta mutazioni nel gene PDS. Queste mutazioni sono ripartite nei 21 esoni del gene. Quattro di esse sono particolarmente frequenti, presenti sul 74% dei cromosomi trasferiti (Blons H., Coyle B.,). Mutazioni della PD S sono evidenziate nella grande maggioranza dei pazienti che presentano una sindrome di Pendred in Francia ed è sempre presente una dilatazione dell’acquedotto del vestibolo nei soggetti in cui sono identificate le due mutazioni(Blons H.).
APPROFONDIMENTO
|
|
|

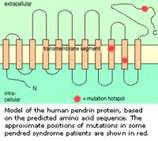
Il gene SLC26A4 (SoLute Carrier; secondo altre fonti SLC viene da Solute Linked Carrier) costituito da 21 esoni, porta quindi alla formazione di una proteina, detta Pendrina, di 780 ami-noacidi, localizzata nella membrana cellulare (detta pertanto transmembrana) con la specifica funzione di trasportare soluti con carica negativa e nella fattispecie iodio (I-), cloro (Cl-) e bicarbonato (HCO3-); è detta anche trasportatore cloro/iodio sodio-indipendente e fa parte della famiglia 26 che ha il nome più generico di “scambiatori anio-nici multifunzione”.
Il gene, localizzato sul braccio lungo del cromosoma 7 (7q21-34), pur essendo presente nel DNA di tutte le cellule, non è espresso ovunque, cioè la pendrina non si trova in tutte le cellule dell’organismo.
La Pendrina è espressa infatti nella tiroide, nell’orecchio interno e nel rene. Un suo difetto funzionale provoca compromissione nel trasporto di iodio, disfunzione tiroidea variabile e spesso gozzo. Il gozzo e l’ipotiroidismo si manifestano generalmente a partire dall’adolescenza ma possono presentarsi sin dalla nascita oppure apparire tardivamente.

Il gene potrebbe però essere responsabile anche di un tipo sordità isolata con assenza di gozzo e di ipotiroidismo.
Infatti alcuni classificano la condizione di Pendred in due sottotipi: quella sindromica, associata a disfunzione tiroidea e gozzo e quella non sindromica in cui vi è la dilatazione dell’acquedotto vestibolare senza ipertrofia/disfunzione ghiandolare tiroidea.
Nella Sindrome di Pendred, a livello dell’orecchio interno, si determina, con meccanismo diverso, disfunzione/danno delle cellule sensoriali della coclea, dove la pendrina è presente sia nelle membrane delle cellule di Deiters, di Claudius, del legamento spirale, del ganglio spirale e del solco esterno ma anche anche nel vestibolo e nel sacco endolinfatico.
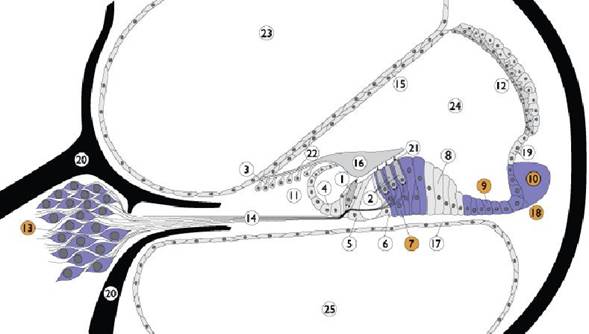
Nella coclea la pendrina è presente nella membrane cellulari del ganglio spirale, del legamento spirale, delle cellule di Claudius, di Dei-ters e del solco esterno. Legenda: 1. Inner hair cell; 2. Outer hair cell; 3. Interdental cells; 4. Inner sulcus cells; 5. Inner pillar cells; 6. Outer pillar cells; 7. Deiter’s cells; 8. Hensen cells; 9. Claudius cells; 10. Spiral ligament; 11. Spiral limbus; 12. Stria vascularis; 13. Spiral ganglion; 14. Auditory nerve; 15. Reissner’s membrane; 16. Tectorial membrane; 17. Basilar membrane; 18. External sulcus cells; 19. Spiral promi-nence; 20. Bony spiral lamina; 21. Reticular lamina; 22. Between IDC and TM; 23. Scala vestibuli; 24. Scala media; 25. Scala tympani

Come può essere trattata la Sindrome di Pendred ?
Le opzioni di trattamento per la sindrome di Pendred sono disponibili. Poiché la sindrome è ereditaria e può coinvolgere tiroide e problemi di equilibrio, molti specialisti possono essere coinvolti nel trattamento, tra cui un medico di base, un audiologo , un endocrinologo , un clinico genetista , un consulente genetico , un otorinolaringoiatra , e un logopedista .
Per ridurre il rischio di perdita dell’udito progressione, i bambini e gli adulti con sindrome di Pendred dovrebbero evitare sport di contatto, che potrebbero portare a lesioni alla testa; indossare una protezione per la testa quando è impegnato in attività come andare in bicicletta e sci, che potrebbe portare a lesioni alla testa; ed evitare situazioni che possono portare a barotrauma (e rapidi sbalzi di pressione), come le immersioni subacquee o il trattamento all’ossigeno iperbarico.
Sindrome di Pendred non è curabile, ma un team di medici lavoreranno insieme per favorire scelte informate circa le opzioni di trattamento. Essi possono anche aiutare le persone a prepararsi per una maggiore perdita di udito e di altre possibili conseguenze a lungo termine della sindrome.
I bambini con sindrome di Pendred devono iniziare il trattamento precoce di acquisire competenze di comunicazione, come imparare il linguaggio dei segni o discorso cued o imparare a usare un apparecchio acustico . La maggior parte delle persone con sindrome di Pendred avranno perdita abbastanza significativa da essere considerati ammissibili per un udito impianto cocleare . Un impianto cocleare è un dispositivo elettronico che viene inserito chirurgicamente nella coclea. Mentre un impianto cocleare non ripristinare o creare un udito normale, si aggira aree danneggiate dell’orecchio per fornire un senso dell’udito nel cervello. I bambini così come gli adulti hanno diritto a ricevere un impianto.
Le persone con sindrome di Pendred che sviluppano un gozzo bisogno di farla controllare regolarmente. Il tipo di gozzo trovato nella sindrome di Pendred è insolito perché anche se cresce in dimensioni, continua ancora a fare la normale quantità di ormone tiroideo. Tale gozzo è spesso chiamato un gozzo eutiroideo.
Syndrome of the month: Pendred Syndrome, J.Med.Genet. 1996;33:1037-1040
http://www.nidcd.nih.gov/health/hearing/pages/pendred.aspx, ( Pendred Syndrome)
http://www.ncbi.nlm.nih.gov/books/NBK1467/, (Pendred Syndrome / DFNB4)
http://www.medicinenet.com/pendred_syndrome/article.htm, (Pendred Syndrome)
Azaiez H, Yang T, Prasad S,et al.,Genotype-phenotype correlations for SLC26A4-related deafnessinHum. Genet., vol. 122, nº 5, December 2007, pp. 451–7,DOI:10.1007/s00439-007-0415-2,PMID17690912.
Coyle B, Coffey R, Armour JA,et al.,Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the nonsyndromic deafness gene DFNB4inNat. Genet., vol. 12, nº 4, April 1996, pp. 421–3,DOI:10.1038/ng0496-421,PMID8630497.
de Moraes VC, dos Santos NZ, Ramos PZ, Svidnicki MC, Castilho AM, Sartorato EL. Molecular analysis of SLC26A4 gene in patients with nonsyndromic hearing loss and EVA: identification of two novel mutations in Brazilian patients, Int J Pediatr Otorhinolaryngol. 2013 Mar;77(3):410-3. doi: 10.1016/j.ijporl.2012.11.042. Epub 2012 Dec 27.
Glaser B., Pendred syndrome. Pediatr Endocrinol Rev. 2003 Dec;1 Suppl 2:199-204; discussion 204. Review
Greinwald J, Dealarcon A, Cohen A, Uwiera T, Zhang K, Benton C, Halstead M, Meinzen-Derr J., Significance of unilateral enlarged vestibular aqueduct, Laryngoscope. 2013 Feb 9. doi: 10.1002/lary.
National Institute on Deafness and Other Communication Disorders,Pendred Syndrome, October 2006.URL consultato il 5 maggio 2008.
Pearce JM, Pendred’s syndrome in Eur. Neurol., vol. 58, nº 3, 2007, pp. 189–90, DOI:10.1159/000104724, PMID17622729
Pendred V,Deaf-mutism and goitreinLancet, vol. 2, nº 3808, 1896, pp. 532,DOI:10.1016/S0140-6736(01)74403-0.
Reardon W, Coffey R, Phelps PD, et al., Pendred syndrome–100 years of underascertainment? (PDF) in QJM, vol. 90, nº 7, July 1997, pp. 443–7, DOI:10.1093/qjmed/90.7.443, PMID9302427.
Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP., The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet. 1999 Apr;21(4):440-3.
Sheffield VC, Kraiem Z, Beck JC,et al.,Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organificationinNat. Genet., vol. 12, nº 4, April 1996, pp. 424–6,DOI:10.1038/ng0496-424,PMID8630498.
Twyffels L, Massart C, Golstein PE, Raspe E, Van Sande J, Dumont JE, Beauwens R, Kruys V.Pendrin: the thyrocyte apical membrane iodide transporter? Cell Physiol Biochem. 2011;28(3):491-6. doi: 10.1159/000335110.
Related citations
Goitrous congenital hypothyroidism and hearing impairment associated with mutations in the TPO and SLC26A4/PDS genes. Pfarr N, Borck G, Turk A, Napiontek U, Keilmann A, Müller-Forell W, Kopp P, Pohlenz J. J Clin Endocrinol Metab. 2006 Jul;91(7):2678-81. Epub 2006 May 9.
www.audiologiainfantile.com Articolo del Dr. Castiglione A.
www.orpha.net
www.xagena.it
www.sin-italy.org
Sindrome di Usher La sindrome Graefe-Usher La sindrome sordità-retinite pigmentosa La sindrome Hallgren
Che cos’è la sindrome di Usher? pag.1
Storia. pag.2
Quanto è diffusa la sindrome di Usher? pag.3
Che cosa provoca la sindrome di Usher? pag.4
Quali sono le caratteristiche dei tre tipi di sindrome di Usher? pag.5
Quali geni sono legati alla sindrome di Usher? pag.7
Come viene diagnosticata la sindrome di Usher? pag.10
Come le persone ereditano la sindrome di Usher? pag.10
È disponibile un test genetico per la sindrome di Usher? pag.10
Che trattamento è disponibile per la sindrome di Usher? pag.11
Implicazioni psicosociali della sindrome di Usher. pag.12
Il ruolo dell’ audiologo. pag.12
Altre forme di ipoacusia associate a perdita della vista. pag.13
Casi individuali .pag.13
Bibliografia. pag.14
Che cos’è la sindrome di Usher? Fig.1
– trasmissione autosomica recessiva
– sordità neurosensoriale congenita (raramente progressiva)
– retinite pigmentosa (RP)
– eterogeneità genetica – Tipi I, Il, III
|
Sindrome di Usher
|
Modalità di trasmissione |
Tipo di deficit uditivo |
Anomalie associate |
||||||
|
⋅⋅⋅ 3-5% delle sordità ⋅⋅⋅ 18% delle retiniti pigmentose ⋅⋅⋅ 50% dei pz sordo-ciechi |
A. R. Eterogeneità genetica (fenomeno per cui mutazioni in loci genetici diversi possono avere lo stesso effetto fenotipico) 12 loci genici e 9 geni identificati come causa di S. di Usher |
|
|
La sindrome di Usher è una condizione genetica che comporta la perdita dell’udito neurosensoriale e retinite pigmentosa (RP) ,è il tipo più comune di perdita di udito autosomica recessiva sindromica. Sebbene sia considerata una malattia rara, è la causa più frequente di sordo-cecità negli esseri umani. Una sindrome è una malattia o un disturbo che ha più di una caratteristica o sintomo. Quando la perdita dell’udito è accompagnata da altri riscontri clinici, è spesso classificato come una sindrome, e ci sono più di 400 tali sindromi descritte da Gorlin, Torell, e Cohen (1995). la sindrome di Usher è una condizione caratterizzata da perdita progressiva dell’udito o sordità e perdita della vista da retinite pigmentosa (RP). Esistono molte forme di s di Usher, ma tre casi su quattro rientrano nel I tipo Questo si presenta con una sordità congenita profonda, areflessia vestibolare bilaterale responsabile di un ritardo motorio di deambulazione (attorno a 18-24 mesi), e retinite che si manifesto più tardivamente, ma sempre nell’infanzia, I primi sintomi visivi consistono in difficoltà visive in penombra, spesso evidenti solo nella seconda decade d’età, ma un esame del fondo oculare ed un elettroretinogramma, possono essere diagnostici anche in età infantile Questa forma richiede una diagnosi precoce, entro i 2-3 anni, perché un impianto cocleare può ridurre di molto le difficoltà comunicative dovute al sommarsi di difetto uditivo e visivo .Nella s di Usher tipo Il la sordità è meno grave, stabile, prevalentemente sulle frequenze acute Anche la retinite interviene più tardivamente ed i difetti vestibolari sono assenti .Nella s di Usher tipo III la sordità è progressiva, mentre i sintomi vestibolari e la retinite pigmentosa insorgono in età variabili .Sono stati localizzarti 11 geni diversi responsabili di queste forme, dei quali otto identificati: 5 per il tipo I, due per il tipo Il, uno per il tipo III. La maggior parte delle s. di Usher sono causate da mutazioni dei geni MYO7A, CDH23, USH2A che codificono per proteine importanti per la costituzione delle cellule cigliate e della matrice extracellulare della coclea. La condizione prende il nome da un oculista britannico, CH Usher, che in un articolo nel 1914 descrisse diversi casi in cui si è sottolineato il legame tra sordità congenita e RP. Tuttavia, nel lontano 1860 lavoratori, come von Graef Lo screening uditivo neonatale ha ridotto l’età di identificazione dei bambini con perdita uditiva 12-18 mesi a 6 mesi o meno (Harrison & Rousch, 1996), ma la diagnosi di sindrome di Usher, con la sua devastante perdita della vista, tipicamente ritardi 5-10 anni dietro l’identificazione della perdita dell’udito (Kimberling & Lindenmuth, 2007). Così, mentre i genitori imparano di perdita uditiva del loro bambino relativamente presto, senza diagnosi differenziale si procede con le decisioni critiche relative all’intervento, comunicazione e opzioni educative ignari che il loro bambino finirà per essere cieco. A causa dello screening uditivo neonatale, audiologi sono spesso contatto primario della famiglia. Pertanto, audiologi sono in grado di migliorare i risultati di diagnosi differenziali per i bambini con sindrome di Usher. Questo articolo è rivolto ad accrescere la comprensione audiologi ‘della presentazione audiologico e visivo, criteri diagnostici e strategie di intervento coinvolti con la sindrome di Usher.
Storia
La sindrome di Usher è chiamato dopo l’oculista scozzese Charles Usher , che ha esaminato la patologia e la trasmissione di questa malattia nel 1914 sulla base di 69 casi. Tuttavia, è stato descritto nel 1858 da Albrecht von Gräfe , (un pioniere della moderna oftalmologia) e Liebreich a Berlino erano a conoscenza del legame tra sordità congenita e RP, soprattutto nei matrimoni consanguinei.. Egli ha riferito il caso di un paziente sordo con retinite pigmentosa , che aveva due fratelli con gli stessi sintomi. Tre anni più tardi(1861), uno dei suoi studenti, Richard Liebreich , ha esaminato la popolazione di Berlino che avevano sordità con retinite pigmentosa. Liebreich ha notato che la sindrome di Usher era di tipo recessivo, dal momento che i casi di combinazioni cieco sordità si sono verificati in particolare nei fratelli dei matrimoni consanguinei o in famiglie con pazienti in diverse generazioni. Le sue osservazioni hanno fornito le prime prove per la trasmissione accoppiata di cecità e sordità, dal momento che non potrebbe essere trovati casi isolati delle due patologie negli alberi genealogici.
La perdita della vista è causata da una malattia dell’occhio chiamata retinite pigmentosa (RP), che colpisce lo strato di tessuto sensibile alla luce nella parte posteriore dell’occhio (retina). Perdita della vista si verifica in quanto le cellule della retina che rilevano la luce si deteriorano gradualmente. Di solito, i bastoncelli della retina sono i primi ad essere interessati, con conseguente precoce cecità notturna e la graduale perdita di visione periferica. In altri casi, si verifica, la degenerazione precoce dei coni della macula, portando ad una perdita di acuità centrale. In alcuni casi, è risparmiata la visione foveale, portando ad una “visione a ciambella”; visione centrale e periferica sono intatti, ma un anello esiste intorno alla regione centrale in cui visione è compromessa . Inizia prima la perdita della visione notturna, cecità notturna, questo disturbo può anche essere accoppiato alla difficoltà ad adattarsi alla luce intensa od a rapidi cambiamenti delle condizioni di luce, seguita da punti ciechi che si sviluppano nella visione laterale (periferica). Nel corso del tempo, questi punti ciechi si ingrandiscono e si fondono per produrre una visione a tunnel. In alcuni casi di sindrome di Usher, la visione è ulteriormente compromessa dalla opacità del cristallino dell’occhio (cataratta). Molte persone con retinite pigmentosa mantiene una certa visione centrale per tutta la vita, però.
Quanto è diffusa la sindrome di Usher?
Si pensa che la sindrome di Usher possa essere responsabile dal 3 per cento, al 6 per cento di tutta la sordità infantile e circa del 50 per cento dei casi di sordità-cecità negli adulti. Si stima che la sindrome Usher di I tipo colpisce almeno 4 individui ogni 100.000 persone. Negli Stati Uniti, i tipi 1 e 2 sono i tipi più comuni. Insieme, essi rappresentano circa il 90 al 95 per cento di tutti i casi di bambini che hanno la sindrome di Usher. Può essere ancora più comune in alcune popolazioni etniche, come nelle etnie di ascendenza ebraica Ashkenazi (dell’Europa centrale e orientale) e nella popolazione Acadian in Louisiana. Si pensa che la sindrome di Usher di II Tipo sia la forma più comune, anche se la frequenza di questo tipo è sconosciuto. Nella maggior parte delle popolazioni la sindrome di Usher di Tipo III rappresenta solo una piccola percentuale di tutti i casi di sindrome di Usher. Questa forma della malattia ,tuttavia, è più comune nella popolazione finlandese, dove rappresenta circa il 40 per cento di tutti i casi.
I ricercatori hanno individuato tre principali tipi di sindrome di Usher, designati come tipo I, II, e III. Questi tipi si distinguono per la loro gravità e l’età in cui i segni ei sintomi compaiono. Il tipo I è ulteriormente suddivisa in sette distinti sottotipi, indicati come tipi IA/IG. La sindrome di Usher di tipo II ha almeno tre sottotipi descritti, designati come tipi IIA, IIB e IIC.
|
|
|
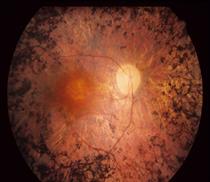
Fig. 1:Fotografia della retina di un paziente con la sindrome di Usher (a sinistra) rispetto ad una retina normale (a destra). Il nervo ottico (freccia) sembra molto chiaro, i vasi (stelle) sono molto sottili e non vi è pigmento caratteristico, chiamato spicole ossee (doppie frecce).
|
|
|
|
Le malattie genetiche possono essere causate da un cambiamento (s) in un gene. Ogni individuo ha due copie dello stesso gene. Malattie genetiche vengono ereditate in modi diversi. La sindrome di Usher è una malattia recessiva.Fig.2a/b Mezzi Recessiva:
Un individuo con la sindrome di Usher solito:
Un individuo che ha una cambiata Usher gene sindrome è chiamato un vettore. Quando due portatori dello stesso gene sindrome di Usher hanno un figlio insieme, con ogni nascita c’è un:
|


Che cosa provoca la sindrome di Usher?
Sindrome di Usher è ereditata, il che significa che si passa dai genitori ai figli attraverso i geni. Geni sono localizzati in quasi ogni cellula del corpo. I geni contengono le istruzioni che dicono le cellule cosa fare. Ogni persona eredita due copie di ogni gene, uno da ciascun genitore. A volte i geni sono alterati o mutati. Geni mutati possono indurre le cellule a agire in modo diverso rispetto al previsto.
La sindrome di Usher è ereditata come carattere autosomico recessivo. Il termine autosomica significa che il gene mutato non è situato su uno dei cromosomi che determinano il sesso di una persona; in altre parole, maschi e femmine possono avere la malattia e può passarlo insieme ad un bambino. La parola recessiva significa che, per avere la sindrome di Usher, una persona deve ricevere una forma mutata del gene sindrome di Usher da ciascun genitore. Se un bambino ha una mutazione in un gene (sindrome Usher), ma l’altro gene è normale, si prevede che avrà un udito ed una visione normale ‘. Le persone con una mutazione in un gene che può causare una malattia autosomica recessiva sono chiamati vettori, perché “portano” il gene con una mutazione, ma non mostrano sintomi del disturbo. Se entrambi i genitori sono portatori di un gene mutato per la sindrome di Usher, avranno una probabilità su quattro di avere un figlio con la sindrome di Usher con ogni nascita.
Di solito, i genitori che hanno un udito normale e la visione non sanno se sono portatori di una mutazione del gene (sindrome di Usher). Attualmente, non è possibile determinare se una persona che non ha una storia familiare di sindrome di Usher è un vettore.
Quali sono le caratteristiche dei tre tipi di sindrome di Usher?
Tipo 1
I bambini con diabete di tipo 1, e sindrome di Usher nascono completamente sordi ( Fig. 3a) o perdono gran parte della loro udito entro il primo anno di vita ed hanno gravi problemi di equilibrio. La perdita progressiva della vista ,causata dalla retinite pigmentosa, si manifesta durante l’infanzia. Questo tipo di sindrome di Usher comprende anche problemi dell’orecchio interno che influenzano l’equilibrio. I genitori dovrebbero consultare il proprio medico e altri professionisti della salute dell’udito il più presto possibile per determinare il miglior metodo di comunicazione per il loro bambino. L’intervento dovrebbe essere introdotto presto, durante i primi anni di vita, in modo che il bambino può approfittare della finestra unica di tempo durante il quale il cervello è più ricettivo per l’apprendimento delle lingue, sia parlato o firmato. Se un bambino viene diagnosticato con il tipo 1, sindrome di Usher presto, prima che lui o lei perde la capacità di vedere, quel bambino ha maggiori probabilità di beneficiare di tutta la gamma di strategie di intervento che possono aiutare lui o lei di partecipare più pienamente nelle attività della vita.
La funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher Tipo I ,a causa dei problemi di equilibrio associati alla sindrome di Usher di tipo 1, i bambini con questo disturbo di solito associato a areflessia vestibolare e ritardo nelle tappe dello sviluppo, come nel controllo del capo e nell’acquisizione della stazione seduta e della deambulazione autonoma, sono lenti a sedersi senza supporto e di solito non camminano autonomamente prima dei 18 mesi di età(Moller, Kimberling, Davenport, Priluck, & White, 1989). Questi bambini di solito iniziano a sviluppare problemi di visione nella prima infanzia, quasi sempre dal momento in cui raggiungono 10 anni. Questa forma richiede una diagnosi precoce, entro i 2-3 anni, perché un impianto cocleare può ridurre di molto le difficoltà comunicative dovute al sommarsi di difetto uditivo e visivo
Problemi di visione più spesso iniziano con difficoltà a vedere di notte, Cecità notturna è tipicamente appare a 10 anni, quindi questi bambini possono avere paura del buio e sono spesso descritti come goffi perché urtano, o inciampare. Il deterioramento significativo del campo visivo e l’acutezza inizia tra la seconda e la terza decade di vita, con la cataratta essendo una complicanza comune (Edwards, Fishman, Anderson, Boschetto, e Derlackie, 1998; Piazza, Fishman, Sugata, Derlacki, e Anderson, 1986; A. Sadeghi, Eriksson, Kimberling, Sjostrom, e Moller, 2006) ma tendono a progredire rapidamente fino a quando la persona è completamente cieco.
|
|
|
Fig. 2a. Tipici risultati audiometrici nella sindrome di tipo 1 Usher.
|

Tipo 2
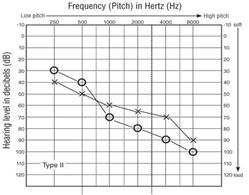
La sindrome di Usher di tipo 2 (circa il 60% dei casi), è caratterizzata da perdita dell’udito dalla nascita Fig.2b ,(la sordità è meno grave, stabile, prevalentemente sulle frequenze acute) e progressiva perdita della vista che inizia in adolescenza o in età adulta e solitamente il quadro è meno grave rispetto al 1 tipo, non associata a alterazioni vestibolari,. La perdita di udito associata a questa forma di sindrome di Usher varia da una forma lieve ad una grave e colpisce soprattutto i toni alti. I bambini affetti hanno problemi di udito, suoni del linguaggio morbido elevate, come quelle delle lettere S e t. Il grado di perdita dell’udito varia all’interno e tra le famiglie con questa condizione. A differenza di altre forme di sindrome di Usher, le persone con diabete di tipo II non hanno problemi di equilibrio causata da problemi dell’orecchio interno.
|
|
|
Figura 2b. Tipici risultati audiometrici di tipo II sindrome di Usher.
|
Tipo 3
I bambini con sindrome di Usher tipo 3 (<3% dei casi, diffusa soprattutto nelle popolazioni Finlandesi e negli Ebrei Ashkenaziti), hanno un udito normale alla nascita, che progredisce durante l’adolescenza ,dopo lo sviluppo del linguaggio, richiedono apparecchi acustici con l’età adulta, la sordità è progressiva, l’ipoacusia è simile a quella descritta per il tipo II ,ma compare attorno ai 3-5 anni e presenta una progressività nel corso degli anni (Kumar et al.. 1984).la perdita dell’udito e perdita della vista sono progressivi a partire dai primi decenni di vita a differenza delle altre forme di sindrome di Usher. Sebbene la maggior parte dei bambini con il disturbo hanno un equilibrio normale o quasi normale, alcuni possono sviluppare problemi di equilibrio in seguito, la disfunzione vestibolare si ha nella metà dei casi, i pazienti presentano un quadro variabile caratterizzato da progressivo peggioramento (Fishman. 1979: Fishman et al.. 1983). Con la mezza età, gli individui più colpiti sono sordi. Anche la perdita della vista causata dalla retinite pigmentosa si sviluppa nella tarda infanzia o nell’adolescenza. Le persone con sindrome di Usher di tipo III possono anche avere difficoltà con l’equilibrio a causa di problemi dell’orecchio interno. Questi problemi variano tra gli individui affetti, Nella s di Usher tipo III i bambini hanno un udito normale alla nascita, mentre i sintomi vestibolari e la retinite pigmentosa insorgono in età variabili
|
Quali geni sono legati alla sindrome di Usher?
Mutazioni nel CDH23, CLRN1, GPR98, MYO7A, PCDH15, USH1C, USH1G, ei geni USH2A causano la sindrome di Usher.
I geni legati alla sindrome di Usher forniscono istruzioni per fare proteine che svolgono ruoli importanti nella udito normale, equilibrio, e la visione.Funzionano nello sviluppo e nel mantenimento delle cellule ciliate, che sono cellule sensoriali dell’orecchio interno che aiutano a trasmettere segnali di movimento del suono e al cervello. Nella retina, questi geni sono coinvolti nel determinare la struttura e la funzione delle cellule sensibili alla luce bastoncelli e coni. In alcuni casi, l’esatto ruolo di questi geni in ascolto e visione è sconosciuta. La maggior parte delle mutazioni responsabili della sindrome Usher portare ad una perdita di cellule ciliate nell’orecchio interno e una graduale perdita di coni e bastoncelli della retina. La degenerazione di queste cellule sensoriali provoca perdita di udito, problemi di equilibrio, e la perdita di visione caratteristica di questa condizione.
Usher sindrome tipo che posso derivare da mutazioni nel CDH23, MYO7A, PCDH15, USH1C, o geneUSH1G. Almeno altri due geni identificati anche causare questa forma di sindrome di Usher.
La sindrome di Usher di tipo II è causata da mutazioni in almeno quattro geni. Solo due di questi geni,USH2A e GPR98 (chiamato anche VLGR1), sono stati identificati.
Le mutazioni in almeno due geni sono responsabili della sindrome di Usher tipo III; tuttavia, CLRN1 è l’unico gene che è stato identificato.
Per saperne di più sul CDH23 , CLRN1 , GPR98 , MYO7A , PCDH15 , USH1C , USH1G , e USH2Ageni.
Nella sindrome di Usher tipo I sono stati identificati 7 sedi di linkage e 4 geni (MYO7A. USH1C. CDH23. PCDH15) a carico dei quali sono state osservate varie mutazioni, con sordità congenita profonda, areflessia vestibolare bilaterale responsabile di un ritardo della deambulazione (deambulazione dopo 18 mesi) e retinite che si sviluppa durante l’infanzia. I primi segni visivi sono dei disturbi della visione in penombra, spesso verso l’inizio del secondo decennio di vita, ma l’esame sistematico del fondo dell’occhio può consentire la diagnosi molto prima di questa età, fin dai 3-4 anni. L’esame più precoce è l’elettroretinogramma, patologico prima dei primi segni nel fondo dell’occhio. La sindrome di Usher di tipo I è un’indicazione all’impianto cocleare precoce per ottenere una comprensione del linguaggio senza lettura labiale in questi bambini che, in età adulta, avranno una rilevante compromissione visiva. Porre la diagnosi attraverso l’esame del fondo dell’occhio a 4 anni è quindi già una situazione tardiva. In linea di principio l’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi, e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale La funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher Tipo I ,da normoreflessia nella sindrome di Usher Tipo II e da risposte che vanno progressivamente peggiorando nella sindrome di Usher Tipo III (Kumar et al. 1984: Möller et al.. 1989)
Nella sindrome di Usher tipo II sono state osservate varie mutazioni a carico dii gene (USH2A); tuttavia è verosimile che vi sia una eterogeneità genetica dal momento che sono state identificate 2 ulteriori sedi di linkage, la sordità è in media grave, non progressiva, predominante sulle frequenze acute, la retinite un poco più tardiva e i segni vestibolari assenti. Nella Usher tipo III è stato recentemente individuato 1 gene (USH3) a carico del quale sono state osservate varie mutazioni (Tab. III). la sordità è progressiva, i segni vestibolari e l’età di inizio della retinite sono variabili (per una rassegna, vedi [Grøndahl J (1987). 8]).
A tutt’oggi sono stati localizzati 12 geni diversi responsabili di queste forme Geni e le proteine che codificano sono stati identificati per 7 dei 12 loci., cinque per la sindrome di Usher tipo I, due per il tipo II e uno per il tipo III (Tabella II/III). MYO7A e CDH23 rappresentano rispettivamente il 30 e 29% dei casi di Usher tipo I e USH2A il 40% dei tipi II(Ouyang X.) La diagnosi molecolare non è eseguita di routine e la diagnosi è essenzialmente clinica.
I geni che causano la sindrome di Usher sono MY07A, USH1C, CDH23, PCDH15,e SANS, che causa USH1, USH2A (che causa USH2 ), e USH3A (che provoca USH3 ). Una mutazione, denominata R245X, del PCDH15 gene può rappresentare una grande percentuale di USH1 casi in Ashkenazi popolazione ebraica di oggi La maggior parte delle s. di Usher sono causate da mutazioni dei geni MYO7A, CDH23, USH2A che codificano per proteine importanti per la costituzione delle cellule cigliate e della matrice extracellulare della coclea.
|
|
|
|
|
|


Tab, II. Genetica della s, di Usher.
|
Locus |
|
Gene |
Marker |
Most lmportant Reference |
OMIM |
|
USH1A |
14q32 |
Unknown |
D14S250, D14S260, D14S292, D14S78 |
Kaplan et al., 1992 |
276900 |
|
USH1B |
11q135 |
MYO7A |
Di 1S906, Dl 1S911, D11S52, OMP-CA |
Weil et al,1995 |
276903 |
|
USH1C |
hp15.1 |
USH1C |
D11S902,D11S921, D11S899,D11S861 |
Smith et al,, 1992 |
276904 |
|
USH1D |
10q |
CDH23 |
D10S529, D10S202, D10S573 |
Wayne et al,, 1996 |
601067 |
|
USH1E |
21q |
Unknown |
D21S1884, |
Chaib et al, 1997 |
602097 |
|
USH1F |
10q2122 |
PCDH15 |
D10S199, D10S578, D10S596 |
Ahmed et aL, 2001 |
602083 |
|
USH1G |
17q24-25 |
Unknown |
|
Mustapha et al.., 2002 |
|
|
USH2A |
1q41 |
USH2A |
D1S229, D1S490, D1S237, D1S474 |
Kimberling et al, |
276901 |
|
USH2B |
3p2324..2, |
Unknown |
D3S1578, D3S3647, D3S3658 |
Hmani et al,, 1999 |
276905 |
|
USH2C |
5q14.3-q21.3 |
Unknown |
D5S428, D5S421 |
Pieke-Dahl et al., 2000 |
605472 |
|
USH 3 |
3q21-q25 |
USH3 |
D3S1299, D3S1555, D3S1280, D3S1 279 |
Sankila et al, 1995 Joensuu et al., 2001 |
276902 |
Geni associati con la sindrome di Usher Tab, III.
|
Tabella 1: geni collegati alla sindrome di Usher |
||||||||
|
Tipo |
Freq [16] |
Gene locus |
Gene |
Proteina |
Funzione |
Dimensioni (AA) |
UniProt |
|
|
USH1B |
39-55% |
11q 13.5 |
Myosin VIIA |
2215 |
||||
|
USH1C |
6-7% |
11p 15.1-p14 |
552 |
|||||
|
USH1D |
19-35% |
10q 21-q22 |
3354 |
|||||
|
USH1E |
raro |
21q 21 |
? |
? |
? |
? |
? |
|
|
USH1F |
11-19% |
10q 11.2-Q21 |
1955 |
|||||
|
USH1G |
7% |
17q 24-q25 |
SANS |
461 |
||||
|
USH2A |
80% |
1q 41 |
Usherin |
Transmembrane linkage |
5202 |
|||
|
USH2C |
15% |
5q 14.3-q21.1 |
VLGR1b |
Molto grande GPCR |
6307 |
|||
|
USH2D |
5% |
9q 32-q34 |
Whirlin |
907 |
||||
|
USH3A |
100% |
3q 21-q25 |
Clarin-1 |
Shaping Synaptic |
232 |
|||
Come viene diagnosticata la sindrome di Usher?
Poiché la sindrome di Usher colpisce l’udito, equilibrio, e la visione, la diagnosi del disturbo di solito include la valutazione di tutti e tre i sensi. Misure comportamentali e oggettive del sistema uditivo sono tecniche familiari per l’audiologo. Proprio come con le misure acustiche, la valutazione della funzione visiva può essere comportamentale (cioè, l’acuità e campo visivo) o oggettiva. Una videoelectronistagmografia (VNG) che misura i movimenti involontari degli occhi potrebbe evidenziare dei problemi di equilibrio. I tests obiettivi comprende l’esame diretto della retina, dove un oculista troverà vasi sanguigni scoloriti, un pallore cereo, e grumi di cellule retiniche morte chiamati spicole ossee (Carr & Noble, 1981). Tuttavia, questi risultati fisici non sono evidenti fino a ben dopo che il paziente è sintomatico. La prova definitiva di RP è un elettroretinogramma (ERG). Come una risposta uditivi del tronco encefalico, l’ERG è una risposta evocata ma dai coni e bastoncelli della retina. Poiché il test richiede l’inserimento di una lente a contatto matrice / elettrodo, anestesia generale è necessaria per i bambini, mentre farmaci topici possono essere utilizzati con gli adulti. Anche se un bambino può avere una visione ancora relativamente buona, l’ERG sarà ridotto o assente quando sindrome RP / Usher è presente. Vi è qualche evidenza che mentre la perdita di ampiezza del ERG è simile tra Usher tipo I e tipo II, il ritardo implicito si può distinguere tra i tipi (Seeliger, Zrenner, Apfelstedt-Sylla, e Jaissle, 2001). Un ERG presenta un vantaggio diagnostico distinto che sarà anormale molto prima dei segni fisici di morte cellulare (spicole ossee) che compaiono sulla retina. Questo spiega perché un oculista spesso non fa la diagnosi con un giovane paziente, in quanto il bambino si esibirà bene nel test dell’acuità visiva e le sue retine sotto esame diretto appariranno normali.
Valutazione degli occhi può includere un test del campo visivo per misurare la visione di una persona periferica, un elettroretinogramma (ERG) per misurare la risposta elettrica delle cellule fotosensibili dell’occhio, e un esame della retina per osservare la retina e altre strutture nella parte posteriore l’occhio. La diagnosi precoce della sindrome di Usher è molto importante. Prima che i genitori sanno che il loro bambino ha la sindrome di Usher, prima il bambino potrà iniziare programmi di formazione educativa speciali per gestire la perdita di udito e della vista.
Come le persone ereditano la sindrome di Usher? APPROFONDIMENTO
Questa condizione è ereditata come carattere autosomico recessivo, il che significa che entrambe le copie del gene in ogni cellula hanno mutazioni. I genitori di una persona con una malattia autosomica recessiva portano ciascuno una copia del gene mutato, ma in genere non mostrano segni e sintomi della malattia.
Dove posso trovare informazioni circa la diagnosi o la gestione della sindrome di Usher?
Tali risorse riguardano la diagnosi o il trattamento della sindrome di Usher e possono includere fornitori di trattamento.
È disponibile un test genetico per la sindrome di Usher?
Finora, sono stati trovati 11 loci genetici (un segmento del cromosoma in cui si trova un certo gene) per causare la sindrome Usher, e sono stati individuati nove geni che causano la malattia. Sono:
-
- Tipo 1 La sindrome di Usher: MY07A, USH1C, CDH23, PCDH15, SANS
-
- Tipo 2 La sindrome di Usher: USH2A, VLGR1, whrn
- Tipo 3 sindrome di Usher: USH3A
Con così tanti possibili geni coinvolti nella sindrome di Usher,i test genetici per la malattia non possono sono effettuati in modo capillare. La diagnosi di sindrome di Usher è di solito effettuata attraverso i tests dell’udito, dell’equilibrio, e della visione. Il test genetico per alcuni dei geni identificati è clinicamente disponibile. Per conoscere i laboratori che effettuano test clinici, visitare il sito Web www.GeneTests.org e cercare la directory laboratorio digitando il termine “sindrome di Usher”. Il test genetico per ulteriori geni sindrome di Usher potrebbe essere disponibile attraverso studi di ricerca clinica. Per informazioni sulle sperimentazioni cliniche che includono il test genetico per la sindrome di Usher, visitare il sito Web www.clinicaltrials.gov e digitare il termine di ricerca “sindrome di Usher” o “test genetici Usher”.
Condotta da tenere
L’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale. La diagnosi precoce è molto importante al fine del programma terapeutico-riabilitativo. La prognosi infatti è caratterizzata dalla comparsa di una cecità attorno ai 3 0-40 anni di età che andrà a sommarsi al deficit uditivo. compromettendo notevolmente il grado di autonomia del soggetto
Che trattamento è disponibile per la sindrome di Usher?
Attualmente non esiste un trattamento medico per prevenire, rallentare la progressione della, o inibire la trasmissione di sindrome di Usher. Impianto cocleare è una valida opzione provata per le persone con sindrome di Usher. Gli individui con tipo I sono candidati dalla nascita, mentre quelli con tipo II o III può diventare candidati nel corso del tempo. Qualità significativo di miglioramento della vita sono state dimostrate per i bambini con sordità congenita che ricevono impianti cocleari (Schorr, Roth, e Fox, 2009), e l’impianto è stato dimostrato come benefico per i bambini sordi con una varietà di disabilità associate (Berrettini et al., 2008). Perché la perdita dell’udito nella sindrome di Usher è cocleare in natura, l’impianto cocleare è una raccomandazione ragionevole e ha, di fatto, dimostrato di beneficiare funzionamento uditiva e sociale dei bambini con sindrome di Usher tipo I (Damon, Pennings, Snik, e Mylanus 2006 ; Liu et al, 2008)..
Ad oggi non esiste alcun trattamento per la RP. Molti individui mantengono un piccolo campo visivo utilizzabile anche in quinta o sesta decade di vita. Vi sono prove preliminari che i fattori ambientali e dietetici possono aumentare la longevità di visione utilizzabile. Questi includono proteggere la retina dalla luce ultravioletta indossando 100% UVA / UVB occhiali da sole e mantenere la salute globale ottimale. L’esercizio fisico e la dieta, come ad esempio a base di pesce ricco di omega-3, sono stati anche suggeriti (Berson, 2000). Gli studi sulla vitamina A e acido docosaesaenoico (DHA) trattamenti hanno creato qualche polemica (Massof & Finkelstein, 1993) e fino ad oggi sono stati solo condotti su persone con RP non sindromica e alcuni Usher di tipo II pazienti (Berson et al sentite., 1993, 2004a , 2004b). I pazienti devono essere avvertiti concernente una supplementazione di vitamina perché consumano troppo può causare ipervitaminosi conseguente cecità, mancanza di crescita, e anche la morte. Le donne e le donne incinte che possono diventare incinte non dovrebbero utilizzare integratori come la vitamina A, perché possono causare difetti alla nascita nel feto.
Implicazioni psicosociali della sindrome di Usher
La perdita sensoriale duale, in particolare la perdita progressiva della vista, presenta molteplici sfide per le persone con sindrome di Usher e le loro famiglie. In genere la perdita dell’udito è stata affrontata e le decisioni sono state fatte sulla metodologia di comunicazione. Una cultura identità Deaf può essere stata stabilita prima conoscenza della perdita della vista imminente. I genitori sono spesso sopraffatti dalla seconda diagnosi e credono che dovrebbero proteggere i loro figli da questa informazione fino a tardi. Nel frattempo, il bambino può sospettare qualcosa ed avere ancora più paura perché lui o lei non capisce. Condivisione di informazioni adeguate all’età con i bambini è il miglior approcio. Non hanno bisogno di sentirsi dire che stanno “diventando ciechi”, ma hanno bisogno di sapere che vedono in modo diverso e che ci sono strategie che li aiuteranno a navigare nel loro mondo. Può essere giustificata l’aiuto di UNA consulenza professionale. Vivere con la sindrome di Usher richiede adattamenti per tutta la vita a cambiare lo stato di visione (Miner, 1995) e, in alcuni casi, cambiare le capacità uditive. Come audiologi, non possiamo supporre ci sono professionisti che hanno familiarità con l’impatto della perdita sensoriale duale. La maggior parte degli specialisti della vista non conoscono la perdita dell’udito e di fatto dipendono dall’audito per aiutare i loro pazienti non vedenti. Gli individui con sindrome di Usher sono sempre sordi o con problemi di udito prima che siano ipovedenti o non vedenti.
Il ruolo dell’ audiologo
La diagnosi di una significativa perdita di udito in un bambino può così dominare la nostra attenzione come audiologi che possiamo trascurare le altre condizioni invalidanti. I genitori si affidano a audiologi per informazioni e supporto nel lavoro con i loro bambini che hanno la perdita dell’udito. Infatti, Steinberg e colleghi (2007) hanno identificato le interazioni dei genitori con l’audiologo come tema importante nel loro esame dei racconti dei genitori in materia di test genetici per la perdita dell’udito. Gli autori hanno indicato che le aspettative dei genitori con gli audiologi includono le informazioni riguardanti le risorse, sostegno e orientamento con i rinvii e test, e permettendo ai genitori di adattarsi alle loro nuovi ruoli. In sintesi, “audiologi sono spesso tenuti a consigliare, guidare e aiutare i genitori attraverso le loro decisioni e le scelte” (Steinberg et al., 2007, p. 64). Inoltre, l’American Speech-Language-Hearing Association (ASHA) Linee guida per la valutazione Audiologic dei bambini dalla nascita ai 5 anni di età consiglia di audiologi dovrebbero “, come opportuno, discutere valutazioni speciali aggiuntivi (ad esempio, la genetica, oculistica, sviluppo infantile) con i genitori / tutori ei neonati primario fornitore di cure “(ASHA, 2004, p. 19). Ciò richiede che l’audiologo avere familiarità con l’epidemiologia genetica della sordità e delle risorse di rinvio e di informazione.
Altre forme di ipoacusia associate a perdita della vista
Secondo la Gallaudet Research Institute (2008), il 40% di tutti i bambini con perdita dell’udito neurosensoriale sono noti per avere una o più condizioni invalidanti supplementari. In questo gruppo, il 10% ha problemi di vista non correggibile. Differenziazione tra i bambini ipovedenti e coloro che sono sordo-ciechi è irto di difficoltà dovute alla variabilità dell’udito residuo e della funzione visiva. Perdita della vista, come la perdita dell’udito, varia notevolmente in gravità e può essere misurata come acuità visiva da vicino e lontano, campo visivo periferico, sensibilità al contrasto, visione dei colori, sensibilità all’abbagliamento, e / o la visione notturna. Così come non si può veramente prevedere la competenza comunicativa di un paziente da un audiogramma per i toni- puri, un singolo parametro visivo non predice il funzionamento visivo.
Mentre Usher è la sindrome più frequentemente associato con RP, ci sono altre condizioni che il clinico deve conoscere. La Sindrome di Alport si manifesta con perdita dell’ udito, nefrite progressiva, e chiazze maculari che possono essere fraintesi come RP, mentre la malattia di Refsum si presenta con anosmia, sordità, e vero RP (Hamel, 2006). Altre condizioni come la sindrome di CHARGE, citomegalovirus, toxoplasmosi, e la meningite può causare contemporaneamente perdita dell’udito e della vista. 2008 Conte Child Nazionale dei bambini e dei giovani che sono sordo-ciechi riporta 10.766 bambini di età compresa nascita attraverso 21 anni sono stati identificati come (Consorzio Nazionale sulla sordocecità, 2008) sordo-ciechi. Indipendentemente dalla eziologia, un audiologo deve essere sensibile alla visione e le strategie suggerite qui per lavorare con i pazienti con sindrome di Usher è applicabile a molti altri pazienti con compromesse delle capacità visive.
Casi individuali da wikipendia
Casi individuali [ modifica ]
Una donna di 31 anni, con la sindrome di Usher, Rebecca Alexander, è stato profilato in Marie Claire nel novembre 2007. [21] Dopo la laurea presso la University of Michigan con ottimi voti, Alexander ha continuato alla Columbia University , dove ha conseguito due gradi di master nella sanità pubblica e clinico sociale. Rebecca è un membro attivo della sua comunità, collaborando con diverse associazioni di beneficenza a New York. La dedizione di Rebecca come membro attivo della sua comunità è stata in particolare riconosciuta quando è stata selezionata come una “Comunità Hero” per correre con la torcia olimpica per i Giochi di Atlanta olimpici del 1996 in onore del suo lavoro di volontariato per il progetto Open Hand, un’organizzazione no-profit consegna pasti per le persone che vivono con l’HIV / AIDS nella zona di San Francisco Bay. Rebecca ha ricevuto la sua formazione psicoterapia psicodinamica attraverso l’American Institute di Psicoanalisi. Attualmente lavora in uno studio privato specializzato nel trattamento dei disturbi dell’umore e d’ansia, disturbi alimentari, dipendenze, disabilità, e traumi. Mentre attualmente facilita seminari di gruppo per la Foundation Fighting Blindness durante le conferenze nazionali, Rebecca è anche in procinto di lanciare l’iniziativa Usher III, un’organizzazione no profit dedicata alla scienza e alla ricerca che cerca di trovare una cura per Usher III. Rebecca insegna ciclismo / spin nelle classi interne con un forte seguito in alcune palestre di New York City. È stata descritta sulla Today Show della NBC il 20 marzo 2009, che è stato nominato per un premio Emmy nel settembre 2010. Lei è la sorella di NBC News Nazionale Corrispondente Peter Alexander.
Christine “Coco” Roschaert [22] è una persona conosciuta con la sindrome di Usher. Ha pubblicato un video blog su YouTube, [23] e recentemente è stato l’altoparlante kick-off per la Settimana della prevenzione della sordità presso l’ Università del Vermont . [24]Nel 2006, si è laureata con una laurea in Scienze della Comunicazione alla Gallaudet University ; lì, lei ha fatto lo sciopero della fame come segno di protesta 2006 organizzata dalla Gallaudet Stati Ora Movimento . [25] Roschaert è ora in Nigeria per la fondazione del programma prima sordociechi .
Una web-community, UsherLife , delle persone con sindrome di Usher è stata fondata il 1 ° febbraio 2005 da Nick Sturley. Anche se centrata sulla Gran Bretagna , che offre risorse per tutte le persone con sindrome di Usher. L’organizzazione sta ospitando regolarmente ritrovi in Inghilterra, come il Usher Hood Pub a Nottingham [26] e un viaggio a Brighton Pier. [27] Altre persone con sindrome di Usher hanno inviato video sulla loro vita e le condizioni su YouTube , in particolare . Ginny Paja-Nyholm[28] Nell’ottobre 2007, Candice, una mamma che vive in Texas, ha iniziato a bloggare sulle sue due figlie, Jasmine e Rebecca;Rebecca che hanno la sindrome di Usher I. [29]
Catherine Fischer ha scritto un’autobiografia ben accolta di “crescere con la sindrome di Usher in Louisiana”, intitolato Orchidea del Bayou. [30] Allo stesso modo, Vendon Wright ha scritto due libri che descrivono la sua vita con la sindrome di Usher, ero cieco, ma ora vedo [31] e attraverso i miei occhi. [32] Louise Boardman ha anche scritto un breve libro intitolato Mio figlio ha la sindrome di Usher. [33]
Christian Markovic, un artista che vive con la sindrome di Usher, gestisce una società, Fuzzy disegni wuzzy. [34]
Spencer Tracy figlio s ‘Giovanni era una persona ben conosciuta con la sindrome di Usher, che ha vissuto una vita piena. [35] IlJohn Tracy Clinic è stata fondata nel 1942 da sua madre Louise per offrire assistenza gratuita ai genitori dei bambini udenti e bambini in età prescolare . [1]
Jacob Desormeaux, figlio del fantino di ippica Kent Desormeaux , ha la sindrome di Usher. Jacob è nato sordo e sta progressivamente diventando cieco. Kent ha dedicato la sua corsa nelle Belmont Stakes , che lui e il suo cavallo darebbero Big Brown la Triple Crown, a suo figlio Giacobbe. La famiglia ha avviato un’organizzazione per raccogliere fondi e la consapevolezza della malattia. La sindrome di Usher è sproporzionatamente comune tra i cajun della Louisiana a sud, come Desormeaux e Fischer, a causa di una mutazione genetica tra i primi francesi Acadian coloni in Nova Scotia .
Elica del DNA co-scopritore e premio Nobel James D. Watson ha pubblicato[36] USH1B mutazioni omozigoti, secondo il suo genoma. Non è chiaro il motivo per cui egli non ha sviluppato la sindrome. Questa mancanza di penetranza genetica sostiene che l’espressione del fenotipo della sindrome di Usher può essere più complessa di quanto inizialmente ipotizzato.
Il “Nalaga’at” israeliano (fare touch) sordo-ciechi Acting Ensemble è composto da 11 attori sordo-cieco, molti dei quali sono diagnosticati con sindrome di Usher. Il gruppo teatrale ha messo su diverse produzioni ed è apparso sia localmente che in Israele e all’estero a Londra e Broadway. [37]
References
Alexander R, Grossman AJ (November 2007). “out of sight, out of sound”. Marie Claire 14 (11): 191–193.American Speech-Language-Hearing Association. (2004). Guidelines for the audiologic assessment of children from birth to 5 years of age. Available from www.asha.org/policy .
Bell J (1933). Retinitis Pigmentosa and Allied Diseases (2nd ed.). London: Cambridge University Press.
Berrettini, S., Forli, F., Genovese, E., Santarello, R., Arslan, E., Chilosi, A., & Cipriani, P. (2008). Cochlear implantation in deaf children with associated disabilities: Challenges and outcomes. International Journal of Audiology, 47, 199–208.
Berson, E. (2000). Nutrition and retinal degeneration. International Ophthalmology Clinics , 40 (4), 93–111.
Berson, E., Rosner, B., Sandberg, M., Hayes, K., Nicholson, B., Weigel-DiFranco, C., & Willett, W. (1993). A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Archives of Ophthalmology, 111 , 761–772.
Berson, E., Rosner, B., Sandberg, M., Weigel-DiFranco, C., Moser, A., Brockhurst, R., et al. (2004a). Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Archives of Ophthalmology, 122 , 1297–1305.
Berson, E., Rosner, B., Sandberg, M., Weigel-DiFranco, C., Moser, A., Brockhurst, R., et al. (2004b). Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Archives of Ophthalmology, 122, 1306–1314.
Boughman, J., Vernon, M., & Shaver, K. (1983). Usher syndrome: Definition and estimate of prevalence from two high-risk populations. Journal of Chronic Diseases, 36, 595–603.
Carr, R., & Noble, K. (1981). Retinitis pigmentosa. Ophthalmology, 88 (2), 169–172.
Chen, D. (2004). Young children who are deaf-blind: Implications for professional in deaf and hard of hearing services. Volta Review, 104 (4), 273–284.
Cohen, M., Bitner-Glindziez, M., & Luxon, L. (2007). The changing face of Usher syndrome: Clinical implications. International Journal of Audiology, 46, 82–93.
Damon, G., Pennings, R., Snik, A., & Mylanus, E. (2006). Quality of life and cochlear implantation in Usher syndrome type I. Laryngoscope, 116, 723–728.
Davenport S, Omenn G (1977). The Heterogeneity of Usher Syndrome (volume 426 ed.). Amsterdam: Excerpta Medica Foundation.
Edwards, A., Fishman, G., Anderson, R., Grove, S., & Derlackie, D. (1998). Visual acuity and visual field impairment in Usher syndrome. Archives of Ophthalmology, 116 , 165–168.
Fishman GA, Kumar A, Joseph ME, Torok N, and Andersonj RJ (1983). “Usher’s syndrome”. Archives of Ophthalmology 101 (9): 1367–1374.
Gallaudet Research Institute. (2008). Regional and national summary report of data from the 2007–08 annual survey of deaf and hard of hearing children and youth. Washington, DC: Author.
Gerber, S; Bonneau, D; Gilbert, B; Munnich, A; Dufier, JL; Rozet, JM; Kaplan, J (2006). “USH1A: chronicle of a slow death”. American Journal of Human Genetics 78 (2): 357–9.
Gorlin R, Tilsner T, Feinstein S, Duvall AJ (1979). “Usher syndrome type III”. Arch. Otolaryngol. 105 (6): 353–354.
Grøndahl J (1987). “Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway”. Clin. Genet. 31 (4): 255–264.
Gorlin, R., Torella, H., & Cohen, M. (1995). Hereditary hearing loss and its syndromes. Oxford, England: Oxford University Press
Hallgren B (1959). “Retinitis pigmentosa combined with congenital deafness with vestibulo-cerebellar ataxia and mental abnormality in a proportion of cases: Clinical and geneto-statistical survey”. Acta Psychiatrica Scandinavica Suppl. 34 (138): 9–101.
Hamel, C. (2006). Retinitis pigmentosa. Orphanet Journal of Rare Diseases, 1 :(40, 1 – 12).
Hammerschlag V (1907). “Zur Kenntnis der hereditaer-degenerativen Taubstummen und ihre differential diagnostische Bedeutung”. Z. Ohrenheilk. 54: 18–36.
Harrison, S., & Rousch, J. (1996). Age of suspicion, identification, and intervention for infants and young children with hearing loss: A national study. Ear and Hearing, 17, 55–62.
Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS (2007). “Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B”. Gene Therapy 14 (7): 584–594
Hicks, W., & Hicks, D. (1981). The Usher’s syndrome adolescent: Programming implications for school administrators, teachers, and residential advisors. American Annals of the Deaf, 26 , 422–431.
Hope CI, Bundey S, Proops D, Fielder AR (1997). “Usher syndrome in the city of Birmingham — prevalence and clinical classification”. British Journal of Ophthalmology 81 (1): 46–53.
Innaccone, A., Kritchevsky, S., Ciccarelli, M., Tedesco, S., Macahuso, C., Kimberling, W., & Somes, G. (2004). Kinetics of visual field loss in Usher syndrome type II. Investigative Ophthalmology & Visual Science, 45 , 784–792.
Keats, B. (2002). Genes and syndromic hearing loss. Journal of Communication Disorders, 33, 355–366.
Kimberling, W., & Lindenmuth, A. (2007). Genetics, hereditary hearing loss, and ethics. Seminars in Hearing, 28 (3), 216–225.
Liebreich R (1861). “Abkunft aus Ehen unter Blutsverwandten als Grund von Retinitis pigmentosa”. Dtsch. Klin. 13: 53.
Liu, X., Angeli, S., Rajput, K., Yan, D., Hodges, A., Eshraghi, A., et al. (2008). Cochlear implantation in individuals with Usher type 1 syndrome. International Journal of Pediatric Otorhinolaryngology, 72, 841–847.
Massof, R., & Finkelstein, D. (1993). Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Archives of Ophthalmology, 111, 751–754.
Merin S, Auerbach E (1976). “Retinitis pigmentosa”. Surv. Ophthalmol. 20 (5): 303–345. doi:10.1016/S0039-6257(96)90001-6
Mets MB, Young NM, Pass A, Lasky JB (2000).”Early diagnosis of Usher syndrome in children”. Transactions of the American Ophthalmological Society 98: 237–45.
Miner, I. (1995). Psychosocial implications of Usher syndrome, type I, throughout the life cycle. Journal of Visual Impairment & Blindness, 89 (3), 287–296.
Moller, C., Kimberling, W., Davenport, S., Priluck, L., & White, V. (1989). Usher syndrome: An otoneurologic study. Laryngoscope, 99, 73–79.
National Consortium on Deaf-Blindness. (2008). National Child Count of Children and Youth Who Are Deaf-Blind . Retrieved December 2, 2009, from www.nationaldb.org/documents/products/2008-Census-Tables.pdf [PDF].
Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C (2001). “A new clinical classification for Usher’s syndrome based on a new subtype of Usher’s syndrome type I”. Laryngoscope 111 (1): 84–86.
Ouyang XM, Yan D, Du LL, Hejtmancik JF, Jacobson SG, Nance WE, Li AR, Angeli S, Kaiser M, Newton V, Brown SD, Balkany T, Liu XZ (2005). “Characterization of Usher syndrome type I gene mutations in an Usher syndrome patient population”. Hum Genet 116 (4): 292–299.
Pakarinen L, Tuppurainen K, Laipapala P, Mäntyjärvi M, Puhakka H (1996). “The ophthalmological course of Usher syndrome type III”. International Ophthalmology 19 (5): 307–311.
Petit, C (2001). “Usher syndrome: from genetics to pathogenesis”. Annual review of genomics and human genetics 2: 271–97.
Piazza, L., Fishman, G., Farer, M., Derlacki, D., & Anderson, R. (1986). Visual acuity loss in patients with Usher’s syndrome. Archives of Ophthalmology, 104 , 1336–1339.
Plantinga, R., Pennings, R., Huygen, P., Sankila, E., Tuppurainen, K., Kleemola, L., et al. (2006). Visual impairment in Finnish Usher syndrome type III. Acta Ophthalmologica Scandinavica, 84 (1), 36–41.
Reiners, J; Nagel-Wolfrum, K; Jürgens, K; Märker, T; Wolfrum, U (2006). “Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease”. Experimental eye research 83 (1): 97–119.
Reisser, C., Kimberling, W., & Otterstedde, C. (2002). Hearing loss in Usher syndrome type II is non progressive. Annals of Otology, Rhinology and Laryngology,111, 1108–1111.
Roux AF, Faugere V, Le Guedard S, Pallares-Ruiz N, Vielle A, Chambert S, Marlin S, Hamel C, Gilbert B, Malcolm S, Claustres M (2006). “Survey of the frequency of USH1 gene mutations in a cohort of Usher patients shows the importance of cadherin 23 and protocadherin 15 genes and establishes a detection rate of above 90%”. J Med Genet 43 (9): 763–768.
Sadeghi, A., Eriksson, K., Kimberling, W., Sjostrom, A., & Moller, C. (2006). Longterm visual prognosis in Usher syndrome types 1 and 2. Acta Ophthalmologica Scandinavica, 84, 537–544.
Sadeghi, M., Cohn, E., Kelly, W., Kimberling, W., Tranebjarg, L., & Moller, C. (2004). Audiological findings in Usher syndrome types IIa and II (non-IIa). International Journal of Audiology, 43, 136–143.
Sadeghi, M., Cohn, E., Kimberling, W., Tranebjarg, L., & Moller, C. (2005). Audiological and vestibular features in affected subject with USH3: A genotype/phenotype correlation. International Journal of Audiology, 44, 307–316.
Sankila EM, Pakarinen H, Kääriäinen H, Aittomäki K, Karjalainen S, Sistonen P, de la Chapelle A (1995). “Assignment of Usher syndrome type III (USH3) gene to chromosome 3q”. Hum. Mol. Genetics 4 (1): 93–98.
Schorr, E., Roth, F., & Fox, N. (2009). Quality of life for children with cochlear implants: Perceived benefits and problems and the perception of single words and emotional sounds . Journal of Speech, Language, and Hearing Research, 52, 141–152.
Seeliger, M., Zrenner, E., Apfelstedt-Sylla, E., & Jaissle, G. (2001). Identification of Usher syndrome subtypes by ERG implicit time. Investigative Ophthalmology and Visual Science, 42, 3066–3071.
Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, et al. (1994). “Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium”. American Journal of Medical Genetics 50 (1): 32–38.
Steinberg, A., Kaimal, G., Ewing, R., Soslow, L., Lewis, K., Krantz, I., & Li, Y. (2007). Parental narratives of genetic testing for hearing loss: Audiologic implications for clinical work with children and families. American Journal of Audiology, 16, 57–67.
Usher C (1914). “On the inheritance of Retinitis pigmentosa with notes of cases”. Roy. Lond. Ophthalmol. Hosp. Rep. 19: 130–236.Wagenaar, M., Snik, A., Kimberling, W., & Cremers, C. (1996). Carriers of Usher syndrome type IB: Is audiometric identification possible? The American Journal of Otology, 17, 853–858.
Vernon M (1969). “Usher’s syndrome — deafness and progressive blindness. Clinical cases, prevention, theory and literature survey”. Journal of Chronic Diseases 22 (3): 133–151.
von Gräfe A (1858). “Exceptionelles Verhalten des Gesichtsfeldes bei Pigmententartung der Netzhaut”. Archiv für Ophthalmologie 4: 250–253.
Wallber, J. (1995–2005). Annual reports of the Eye & Ear Clinic. Rochester, NY: National Technical Institute for the Deaf at Rochester Institute of Technology.
Williams DS (2007). “Usher syndrome: Animal models, retinal function of Usher proteins, and prospects for gene therapy”. Vision Research xx (3): xx–xx.doi:10.1016/j.visres.2007.08.015Web Resources
Boys Town National Research Hospital Usher Syndrome Web page
National Consortium on Deaf-Blindness
National Consortium on Deaf-Blindness, State Deaf-Blind Projects
National Institute on Deafness and Other Communication Disorders Usher Syndrome Web page
National Institutes of Health Usher Syndrome Web page
National Library of Medicine Genetics Home Reference
Second Sight Medical Products, Inc. retinal prosthesis—investigational device
Understanding Usher Syndrome: Information for School Counselors [PDF, 2 MB]
La sindrome Jervell e Lange-Nielsen(JLNS), sindrome autosomica recessiva del QT lungo (LQTS), sindrome della morte improvvisa, La sindrome cardiouditiva, La sindrome cardiouditiva di Jervell e Lange-Nielsen, sordità congenita e malattie cardiache funzionali.
Che cosa è la sindrome Jervell e Lange-Nielsen? pag.1
Cenni storici pag.2
Quanto comune è la sindrome Jervell e Lange-Nielsen? Epidemiologia pag.2
Quali geni sono legati alla sindrome Jervell e Lange-Nielsen? pag.3
Come fanno le persone ereditano sindrome Jervell e Lange-Nielsen?
VARIANTI CLINICHE: Sindrome di Romano-Ward (RWS) (senza deficit uditivo) pag.5
La sindrome di Brugada – La Sindrome del QT lungo acquisita – pag.5
Sindrome di Jervell-Lange-Nielsen (associata a sordità congenita) pag.5
Sintomi pag.6
Diagnosi pag.7
Trattamento pag.7
APPROFONDIMENTI pag.11/33
SINDROME DEL QT LUNGO pag.33/64
Che cosa è la sindrome Jervell e Lange-Nielsen?
Sindrome di Jerwell e Lange-Nielsen(JLNS)
– trasmissione autosomica recessiva
– sordità neurosensoriale congenita
– difetti ritmo cardiaco, prolungato Q-T
– elevato rischio di morte improvvisa
|
Sindrome di Jervell-LangeNielsen
|
Modalità di trasmissione |
Tipo di deficit uditivo |
Anomalie associate |
|
1% delle sordità |
A. R. gene KCNQ1, KCNE1 |
Sordità neurosensoriale, prelinguale grave-profonda |
Anomalie cardiache (Q-T lungo>460-480 msec.) Torsione di punta, sincope e talora morte improvvisa |
La sindrome di Jervell e Lange-Nielsen (JLNS) è una variante autosomica recessiva della sindrome familiare del QT lungo (LQTS, vedi sotto), caratterizzata da sordità neurosensoriale bilaterale profonda congenita, è il terzo tipo più comune di perdita di udito autosomica sindromica, intervallo QT lungo all’elettrocardiogramma e tachiaritmie ventricolari. In questa forma sono stati identificati due geni KCNQ1 e KCNE1 che normalmente codificano per proteine-canale specifiche per il potassio, a livello della stria vascolare. Benché questa sindrome abbia una bassa prevalenza (1 /100.000), la sua diagnosi è importante, perché gli episodi di morte improvvisa per arresto cardiaco possono essere prevenuti Essa può essere facilmente individuata quando ad una sordità congenita si associa un’anomalia elettrocardiografica. Anomalie ECG possono anche individuare portatori eterozigoti normoudenti. Va prestata molta attenzione ai farmaci che causano un allungamento del Q-T, dato il difetto congenito del ritmo cardiaco.
|
|
|
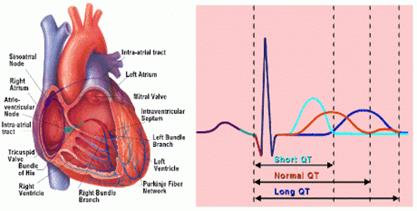

Cenni storici
Fu riscontrata per la prima volta nel 1957 da Jervell e Lange-Nielsen, che descrissero la variante della sindrome associata a sordità.[ Jervell A, Lange-Nielsen F., 1957] Hanno fornito la prima descrizione completa del problema di conduzione elettrica nel cuore chiamato sindrome del QT lungo (LQTS). LQTS si riferisce alla QT misurata sul elettrocardiogramma che indica che il muscolo cardiaco richiede più tempo del solito per ricaricare tra i battiti. Esso predispone le persone colpite da tachiaritmie chiamati torsione di punta (TdP), che porta alla sincope e può causare morte cardiaca improvvisa I due autori notarono che gli episodi di morte improvvisa avevano carattere familiare (si trasmetteva nei discendenti un aumentato rischio). Successivamente, nel 1958, Levine e Woodworth osservarono una morte improvvisa di un tredicenne con i sintomi tipici della malattia.[ Levine SA, Woodworth CR., 1958] Negli anni ’60 due studi indipendenti, l’uno condotto da Romano nel 1963 e l’altro da Ward nel 1964, contribuirono alla definizione di una sindrome, successivamente denominata sindrome di Romano-Ward, caratterizzata da attacchi sincopali ed anomalie dell’ECG, senza deficit uditivo.
Quanto è comune la sindrome Jervell e Lange-Nielsen? Epidemiologia
La sindrome Jervell e Lange-Nielsen è raro; essa colpisce circa 1,6-6 per 1 milione di persone in tutto il mondo. Questa condizione ha una prevalenza più elevata in Danimarca, Norvegia e la Svezia dove colpisce almeno 1 su 200.000 persone. La sindrome colpisce prevalentemente le giovani donne sembrano avere un minor numero di eventi potenzialmente letali. JLNS è generalmente presente nella prima infanzia con una probabilità del 90% dei problemi sintomatici dall’età di 18. La prevalenza della malattia è di 1 a 6 persone per 1.000.000 nati vivi. Fra i soggetti affetti, l’incidenza di torsione di punta, sincope e morte cardiaca improvvisa è più elevata in caso di sordità congenita, precedente tachiaritmia o sincope; la stessa aumenta inoltre proporzionalmente all’allungamento dell’intervallo QT. Si calcola che il rischio relativo di aritmie maligne aumenti di 1,1 – 1,2 volte per ciascun prolungamento di 10 msec del QTc oltre i valori normali.[ Hurst, 2005.] È rara (1/100.000), ma è molto più comune nei paesi a elevata frequenza di matrimoni consanguinei. Circa il 50% dei pazienti diventa sintomatico prima dei tre anni. ma di diagnosi facile tramite un elettrocardiogramma (ECG) sistematico in caso di sordità grave o profonda congenita(Splawski I.). L’ECG mostra un allungamento dell’intervallo QT JLNS, che è di solito molto allungato (>500 ms) e si associa a tachiaritmie (compresa la tachicardia ventricolare, episodi di tachicardia ventricolare a torsioni di punta [TdP] e fibrillazione ventricolare) che possono causare sincope o morte improvvisa. La JLNS è una variante molto grave della LQTS. I pazienti diventano sintomatici molto prima, rispetto a quelli affetti dalle altre forme della LQTS. Circa il 90% dei pazienti presenta accidenti cardiaci scatenati da emozioni intense e improvvise, competizioni sportive, paura o tuffi nell’acqua fredda che traduce un disturbo della conduzione cardiaca, fonte di malessere o di morte improvvisa che possono manifestarsi senza circostanze scatenanti o in seguito a uno stress.
Quali geni sono legati alla sindrome Jervell e Lange-Nielsen?
Le mutazioni nei geni KCNQ1 KCNE1 e causano la sindrome Jervell e Lange-Nielsen.
I geni KCNE1 e KCNQ1 forniscono istruzioni per fare proteine che lavorano insieme per formare un canale attraverso le membrane cellulari. Questi trasporti canali caricata positivamente atomi di potassio (ioni) di cellule. Il movimento di ioni potassio attraverso questi canali è fondamentale per mantenere le normali funzioni di strutture dell’orecchio interno e muscolo cardiaco.
La JLNS è dovuta alle mutazioni eterozigoti composte o omozigoti del gene KCNQ1 (locus LQT1; 11p15.5) e/o del gene KCNE1 (locus LQT5; 21q22.1-q22.2) ed è trasmessa come carattere autosomico recessivo.. La prevenzione è possibile mediante un trattamento medico. Due geni sono stati identificati, KCNQ1 e KCNE1, (Splawski I., Neyroud N., Wang Z., Chen Q.) che codificano per alcuni canali del potassio localizzati nella stria vascolare (Tabella 4). Circa il 90% dei casi di sindrome Jervell e Lange-Nielsen sono causate da mutazioni nel gene KCNQ1; le mutazioni KCNE1 sono responsabili per il restante 10% dei casi. Questi due geni producono (codifica) proteine essenziali per la funzione dei canali ionici del cuore e il tubo lumaca definente parte dell’orecchio interno (coclea). Canali ionici regolano il movimento delle particelle elettricamente cariche (ioni esempio, potassio e sodio) in alcune strutture dell’orecchio e del cuore. Questi ioni trasportano gli impulsi elettrici necessari per l’udito e la normale funzione del cuore. Le mutazioni di questi geni provocano funzione anormale dei canali ionici e, a sua volta, influenzano l’udito e il corretto funzionamento del sistema elettrico del cuore JLNS viene ereditata come una malattia genetica autosomica recessiva, causata da mutazioni in entrambe le copie di un gene, quello ricevuto dal padre e uno dalla madre.

Gli investigatori hanno determinato che il gene KCNQ1 si trova sul braccio corto (p) del cromosoma 11 (11p15.5). I cromosomi, che sono presenti nel nucleo delle cellule umane, portano l’informazione genetica per ogni individuo. Coppie di cromosomi umani sono numerate da 1 a 22, e una coppia aggiuntiva di 23 cromosomi sessuali che comprendono un X e un cromosoma Y nei maschi e due cromosomi X nelle femmine. Ogni cromosoma ha un braccio corto designato “p” e un lungo braccio designato “q”. I cromosomi sono ulteriormente suddivisi in molti gruppi che sono numerati. Per esempio, “cromosoma 11p15.5” si riferisce alla fascia 15 sul braccio corto del cromosoma 11. Le bande numerati indicano la posizione delle migliaia di geni che sono presenti su ciascun cromosoma.
The KCNQ1 gene is located on the short (p) arm of chromosome 11 at position 15.5.
Il gene KCNE1 si trova sul braccio lungo (q) del cromosoma 21 (21q22.1-q22.2).
Malattie genetiche recessive si verificano quando un individuo eredita lo stesso gene anomalo per lo stesso tratto da ciascun genitore. Se una persona riceve un gene normale e un gene per la malattia, la persona sarà eterozigote ed un veicolo per la malattia, ma di solito non mostra sintomi. Alcuni portatori di una o KCNQ1 KCNE1 mutazione del gene hanno perturbazione del normale ritmo cardiaco, ma il loro udito è di solito normale. Il rischio per due genitori portatori sia per trasmettere il gene difettoso e, quindi, hanno un figlio affetto è del 25% ad ogni gravidanza. Il rischio di avere un figlio che è un vettore come i genitori è del 50% ad ogni gravidanza. La possibilità per un bambino di ricevere i geni normali da entrambi i genitori e di essere geneticamente normale per quel particolare tratto è del 25%. Tra fratelli e sorelle affetti in un sibship con un disturbo recessivo, il rischio di essere portatore è di 2/3, o il 67%.
I genitori portatori eterozigoti della mutazione possono presentare dei malesseri (donde l’importanza dell’anamnesi familiare) e un’ipoacusia. Alcuni casi di JLNS hanno avuto genitori che erano legati dal sangue (consanguinei). Tutti gli individui portano 4-5 geni anormali. I genitori che sono parenti stretti hanno una probabilità maggiore di genitori non imparentati sia per portare lo stesso gene anomalo, che aumenta il rischio di avere bambini con una malattia genetica recessiva. Benché questa sindrome abbia una bassa prevalenza (1 /1 00.000), La diagnosi si basa sulla sordità neurosensoriale congenita, associata a intervallo QT lungo e mutazioni nei geni-malattia (KCNQ1, KCNE1). I test molecolari sono di supporto alla clinica. Nelle famiglie nelle quali è nota la mutazione patogenetica possono essere consigliati i test prenatali e la diagnosi genetica preimpianto. La diagnosi differenziale della sordità si pone con altre forme, congenite e acquisite, sindromiche e non sindromiche, di sordità neurosensoriale.
VARIANTI CLINICHE
Sintomi delle seguenti disturbi possono essere simili a quelli di JLNS. I confronti possono essere utili per una diagnosi differenziale Si distinguono diverse sindromi caratterizzate da allungamento dell’intervallo QT e rischio di morte improvvisa per aritmie ventricolari:
Sindrome di Romano-Ward (RWS) (senza deficit uditivo) La sindrome di Romano-Ward è una rara malattia cardiaca genetica caratterizzata da anomalie che colpiscono il sistema elettrico del cuore che provocano la sindrome del QT lungo. La gravità della RWS varia notevolmente da caso a caso. Alcuni individui possono non avere sintomi apparenti (asintomatici); altri possono sviluppare in modo anomalo aumento battito cardiaco (tachiaritmie) con conseguente episodi di perdita di coscienza (sincope), arresto cardiaco e morte improvvisa potenzialmente. RWS è ereditata come carattere autosomico dominante. (Per maggiori informazioni su questo disturbo, scegliere “Romano-Ward” come termine di ricerca nel database Malattie Rare.)
La sindrome di Brugada è una rara malattia cardiaca ereditaria caratterizzata da anomalie del sistema elettrico del cuore. I sintomi variano molto da caso a caso. Alcuni individui affetti sperimenteranno sintomi apparenti; altri possono sviluppare sintomi simili a JLNS e RWS con battiti cardiaci irregolari che portano a episodi di perdita di coscienza (sincope), arresto cardiaco, e, potenzialmente, la morte improvvisa spesso durante il sonno. La sindrome di Brugada è ereditata come carattere autosomico dominante. (Per maggiori informazioni su questo disturbo, scegliere “Brugada” come termine di ricerca nel database Malattie Rare.)
La Sindrome del QT lungo acquisita è una malattia cardiaca rara caratterizzata da anomalie del ritmo cardiaco potenzialmente con conseguente perdita di coscienza, arresto cardiaco e morte improvvisa. Il disturbo si verifica più spesso secondaria alla somministrazione di alcuni farmaci. Alcuni ricercatori ritengono che gli individui affetti possono essere geneticamente suscettibilità allo sviluppo della sindrome del QT lungo acquisita. Disturbi neurologici, ictus, e gli squilibri elettrolitici sono stati indicati come possibili cause della sindrome del QT lungo acquisita
Sindrome di Jervell-Lange-Nielsen (associata a sordità congenita)
La diagnosi differenziale della cardiopatia si pone con la sindrome di Romano-Ward e con le altre forme di LQTS (si vedano questi termini), con le anomalie degli elettroliti (ipokaliemia, ipomagnesemia e ipocalcemia), con l’ipotensione ortostatica, la sincope vasovagale e la LQTS indotta dai farmaci. La sordità della JLNS può essere trattata con impianti cocleari. Il principale obiettivo della presa in carico nella JLNS è la prevenzione della sincope, dell’arresto cardiaco e della morte improvvisa. Il trattamento è complicato dall’età precoce dei pazienti nel momento in cui la maggior parte di essi diventa sintomatico. Dato che l’efficacia dei soli farmaci beta-bloccanti è parziale, deve essere preso seriamente in considerazione l’uso del defibrillatore cardioverter impiantabile (ICD). Con l’impiego di terapie complementari (pacemaker, ICD e denervazione simpatetica sinistra), oltre il 50% dei pazienti presenta altri sintomi e rischia di andare incontro a morte improvvisa. I familiari devono essere formati sulla procedura di rianimazione cardiopolmonare, in quanto più del 95% dei pazienti affetti da JLNS soffre di un evento cardiaco prima dell’infanzia. Oltre la metà dei bambini non trattati muore prima dei 15 anni.la sua diagnosi è importante, perché gli episodi di morte improvvisa per arresto cardiaco possono essere prevenuti Essa può essere facilmente individuata quando ad una sordità congenita si associa un’anomalia elettrocardiografica. Anomalie ECG possono anche individuare portatori eterozigoti normoudenti. Va prestata molta attenzione ai farmaci che causano un allungamento del Q-T, dato il difetto congenito del ritmo cardiaco.

Sintomi
I sintomi della sindrome di JLNS (sindrome di Jervell e Lange-Nielsen) sono di solito evidenti durante la prima infanzia. La perdita dell’udito è rilevata alla nascita o nella prima infanzia. La perdita dell’udito associati a JLNS è causata dall’incapacità dei nervi uditivi per trasmettere informazioni sensoriali al cervello (ipoacusia neurosensoriale) e colpisce entrambe le orecchie (bilaterali). In perdita dell’udito JLNS di solito è profonda, ma tende a colpire l’audizione delle alte frequenze più di basse frequenze. Bassi livelli di ferro e di aumento dei livelli di gastrina sono spesso presenti nei pazienti con JLNS, che possano portare ad anemia ferro carente.
Il sintomo cardiaco più comune associato alla JLNS è la perdita parziale o totale della coscienza (sincope o svenimento), accompagnata da ritmi cardiaci anormalmente veloci noto come torsione di punta (TDP). TDPS possono progredire ad una condizione più grave conosciuta come fibrillazione ventricolare, in cui la normale attività elettrica del cuore diventa disordinato con conseguente battito cardiaco non coordinati e malfunzionamento dei principali camere di pompaggio del cuore (ventricoli). Di conseguenza, poca o nessuna sangue viene pompato dal cuore. La fibrillazione ventricolare potenzialmente traduce in arresto cardiaco o morte improvvisa.
I sintomi della JLNS come la sincope tendono a verificarsi senza preavviso ed a ripresentarsi in modo imprevisto. Sforzi eccessivi, eccitazione o stress possono scatenare questi sintomi ricorrenti, anche se spesso iniziano senza una causa particolare, è troppo. In alcuni casi, gli episodi possono essere innescati da “spavento” eventi come una sveglia andare fuori o il telefono che squilla nel cuore della notte. La gravità e la frequenza degli attacchi vari. Alcune persone possono avere dolore toracico lieve senza perdita di coscienza; altri possono perdere coscienza in tutto o avere crisi di grande male seguiti da un periodo di disorientamento. In alcuni casi disturbi possono essere il primo sintomo cardiaco presentazione di JLNS. La gravità e la frequenza degli episodi, spesso diminuiscono durante la mezza età. I sequestri sono spesso mal diagnosticato come epilessia e trattati di conseguenza per diversi anni prima della diagnosi corretta è fatto.
Terapie standard
Diagnosi
Una diagnosi di JLNS viene effettuata sulla base di una valutazione clinica approfondita, una storia dettagliata del paziente e un test specializzato chiamato un elettrocardiogramma (ECG o EKG). I bambini con congenito sordità neurosensoriale, in particolare quelli con una storia inspiegabile di svenimento, sincope o arresto cardiaco improvviso, dovrebbero essere valutati per JLNS. Un elettrocardiogramma registra gli impulsi elettrici del cuore, può rivelare modelli elettrici anomali come ad esempio un prolungato intervallo QT caratteristiche delle persone con JLNS. La diagnosi può essere confermata attraverso test genetici molecolari che individuano le mutazioni malattia-specifici nel gene KCNQ1 o il gene KCNE1. Gene sequenziamento del gene KCNQ1 è di solito eseguita come primo passo poiché la maggior parte individui affetti hanno mutazioni in questo gene.
Trattamento
Il trattamento delle persone con JLNS mira a trattare la perdita di udito e prevenire sintomi caratteristici come la perdita di coscienza o arresto cardiaco. Farmaci specifici, evitare di innescare eventi e determinati dispositivi medici possono tutti essere usati per trattare le persone con JLNS.
La perdita dell’udito in persone con JLNS può essere trattato con un piccolo dispositivo noto come un impianto cocleare. A differenza di aiuti, che aumentano e chiariscono il suono udito, un impianto cocleare migliora l’udito, stimolando le fibre nervose all’interno dell’orecchio interno.
Il trattamento di scelta per le anomalie cardiache nella maggior parte dei soggetti con JLNS è la terapia farmacologica con farmaci beta-adrenergici (beta-bloccanti). I beta-bloccanti, che comprendono propranololo, atenololo, e nadololo, ridurre il carico di lavoro del cuore diminuendo la stimolazione elettrica del cuore.
Individui per i quali i beta-bloccanti hanno successo possono essere trattati con una procedura chirurgica in cui certi nervi che vanno al cuore vengono rimossi (a sinistra denervazione simpatica cardiaca o simpaticectomia). Tuttavia, di recente il trattamento con un sistema automatico defibrillatore cardioverter impiantabile (ICD) ha sostituito simpaticectomia come il trattamento di scelta in questi individui. Questo dispositivo rileva il battito cardiaco anormale automaticamente e selettivamente fornisce un impulso elettrico al cuore. ICD sono utilizzati in combinazione con la terapia farmacologica antiarrythmic. Alcuni individui con JLNS sono incoraggiati ad evitare possibili eventi scatenanti come il salto in acqua fredda, giri del parco di divertimenti o sport competitivi.
La consulenza genetica è raccomandata per le persone colpite e delle loro famiglie. Altro trattamento è sintomatico e di supporto.
Terapie sperimentali
Un dispositivo impiantabile, il pacemaker cibernetico QT-sensibili, è attualmente testato per le persone con JLNS alto rischio. Questa unità può essere in grado di monitorare il ritmo cardiaco e rilevare lo sviluppo di gravi irregolarità del battito cardiaco. Efficacia e gli effetti collaterali di questi dispositivi impiantati non sono stati pienamente documentati e più vasta ricerca viene perseguito prima che il loro valore terapeutico per JLNS può essere valutato. Literature Cited
-
- Ackerman MJ. Cardiac causes of sudden unexpected death in children and their relationship to seizures and syncope: genetic testing for cardiac electropathies. Semin Pediatr Neurol. 2005;12:52–8. [PubMed]
-
- Allan WC, Timothy K, Vincent GM, Palomaki GE, Neveux LM, Haddow JE. Long QT syndrome in children: the value of the rate corrected QT interval in children who present with fainting. J Med Screen. 2001;8:178–82. [PubMed]
-
- Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Schwartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7. [PubMed]
-
- Baek JS, Bae EJ, Lee SY, Park SS, Kim SY, Jung KN, Noh CI. Jervell and Lange-Nielsen syndrome: novel compound heterozygous mutations in the KCNQ1 in a Korean family. J Korean Med Sci. 2010;25:1522–5. [PMC free article] [PubMed]
-
- Berge KE, Haugaa KH, Früh A, Anfinsen O-G, Gjjesdaj K, Siem G, Øyen N, Greve G, Carlsson A, Rognum TO, Hallerud M, Kongsgård E, Amlie JP, Leren TP. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand J Clin Lab Invest. 2008;68:362–8. [PubMed]
-
- Berul CI, Perry JC. Contribution of long-QT syndrome genes to sudden infant death syndrome: is it time to consider newborn electrocardiographic screening? Circulation. 2007;115:294–6. [PubMed]
-
- Bhuiyan ZA, Momenah TS, Amin AS, Al-Khadra AS, Alders M, Wilde AA, Mannens MM. An intronic mutation leading to incomplete skipping of exon-2 in KCNQ1 rescues hearing in Jervell and Lange-Nielsen syndrome. Prog Biophys Mol Biol. 2008;98:319–27. [PubMed]
-
- Broomfield SJ, Bruce IA, Henderson L, Green KMJ, Ramsden RT. Cochlear impantation in Jervell & Lange-Nielsen syndrome: a cautionary report. Cochlear Implants Int. 2010;11:163–5. [PubMed]
-
- Chen Q, Zhang D, Gingell RL, Moss AJ, Napolitano C, Priori SG, Schwartz PJ, Kehoe E, Robinson JL, Schulze-Bahr E, Wang Q, Towbin JA. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome. Circulation. 1999;99:1344–7. [PubMed]
-
- Chorbachi R, Graham JM, Ford J, Raine CH. Cochlear implantation in Jervell and Lange-Nielsen syndrome. Int J Pediatr Otorhinolaryngol. 2002;66:213–21. [PubMed]
-
- Daneshi A, Ghassemi MM, Talee M, Hassanzadeh S. Cochlear implantation in children with Jervell and Lange-Nielsen syndrpome. J Laryngol Otol. 2008;122:314–7. [PubMed]
-
- Duggal P, Vesely MR, Wattanasirichaigoon D, Villafane J, Kaushik V, Beggs AH. Mutation of the gene for IsK associated with both Jervell and Lange- Nielsen and Romano-Ward forms of Long-QT syndrome. Circulation. 1998;97:142–6. [PubMed]
-
- Eddy CA, MacCormick JM, Chung SK, Crawford JR, Love DR, Rees MI, Skinner JR, Shelling AN. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm. 2008;5:1275–81. [PubMed]
-
- Gao Y, Li C, Liu W, Wu R, Qiu X, Liang R, Li L, Zhang L, Hu D. Genotype-phenotype analysis of three Chinese families with Jervell and Lange-Nielsen syndrome. J Cardiovasc Dis Res. 2012;3:67–75. [PMC free article] [PubMed]
-
- Goel AK, Berger S, Pelech A, Dhala A. Implantable cardioverter defibrillator therapy in children with long QT syndrome. Pediatr Cardiol. 2004;25:370–8. [PubMed]
-
- Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, Towbin JA, Ackerman MJ, Murphy L. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006;17:1161–8. [PubMed]
-
- Green JD, Schuh MJ, Maddern BR, Haymond J, Helffrich RA. Cochlear implantation in Jervell and Lange-Nielsen syndrome. Ann Otol Rhinol Laryngol Suppl. 2000;185:27–8. [PubMed]
-
- Hazinski MF, Markenson D, Neish S, Gerardi M, Hootman J, Nichol G, Taras H, Hickey R. Response to cardiac arrest and selected life-threatening medical emergencies: the medical emergency response plan for schools: A statement for healthcare providers, policymakers, school administrators, and community leaders. Circulation. 2004;109:278–91. [PubMed]
-
- Hothi SS, Thomas G, Killeen MJ, Grace AA, Huang CL. Empirical correlation of triggered activity and spatial and temporal re-entrant substrates with arrhythmogenicity in a murine model for Jervell and Lange-Nielsen syndrome. Pflugers Arch. 2009;458:819–35. [PMC free article] [PubMed]
-
- Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell.2001;104:569–80. [PubMed]
- Larsen LA, Fosdal I, Andersen PS, Kanters JK, Vuust J, Wettrell G, Christiansen M. Recessive Romano-Ward syndrome associated with compound heterozygosity for two mutations in the KVLQT1 gene. Eur J Hum Genet. 1999;7:724–8. [PubMed]
22. Levine SA, Woodworth CR., Congenital deaf-mutism, prolonged QT interval, syncopal attacks and sudden death. in N Engl J Med. 1958 ., vol. 28, agosto 1958, pp. 412-417.
-
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet. 1997;15:186–9.[PubMed]
-
- Ning L, Moss AJ, Zareba W, Robinson J, Rosero S, Ryan D, Qi M. Novel compound heterozygous mutations in the KCNQ1 gene associated with autosomal recessive long QT syndrome (Jervell and Lange-Nielsen syndrome). Ann Noninvasive Electrocardiol. 2003;8:246–50. [PubMed]
-
- Ocal B, Imamoglu A, Atalay S, Ercan Tutar H. Prevalence of idiopathic long QT syndrome in children with congenital deafness. Pediatr Cardiol. 1997;18:401–5. [PubMed]
-
- Ohno S, Kubota T, Yoshida H, Tsuji K, Makiyama T, Yamada S, Kuga K, Yamaguchi I, Kita T, Horie M. A novel mutation associated with Jervell and Lange-Nielsen syndrome in a Japanese family. Circ J. 2008;72:687–93. [PubMed]
-
- Olesen MS, Bentzen BH, Nielsen JB, Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen JH, Schmitt N. Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med Genet. 2012;13:24. [PMC free article] [PubMed]
-
- Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, Toivonen L, Kontula K. A founder mutation of the potassium channel KCNQ1 in long QT syndrome: implications for estimation of disease prevalence and molecular diagnostics. J Am Coll Cardiol. 2001;37:562–8.[PubMed]
- Priori SG, Barhanin J, Hauer RN, Haverkamp W, Jongsma HJ, Kleber AG, McKenna WJ, Roden DM, Rudy Y, Schwartz K, Schwartz PJ, Towbin JA, Wilde AM. Genetic and molecular basis of cardiac arrhythmias: impact on clinical management parts I and II. Circulation. 1999;99:518–28.[PubMed]
30. Ray WA, Murray KT, Meredith S, Narasimhulu SS, Hall K, Stein CM, Oral erythromycin and the risk of sudden death from cardiac causes in N. Engl. J. Med., vol. 351, nº 11, 2004, pp. 1089–96, DOI:10.1056/NEJMoa040582, PMID 15356306.
-
- Richter S, Brugada P. Risk-stratifying Jervell and Lange-Nielsen syndrome from clinical data. J Cardiovasc Electrophysiol. 2006;17:1169–71. [PubMed]
- Rivas A, Francis HW. Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol Neurotol. 2005;26:415–24. [PubMed]
33. Romano C, Gemme G, Pongiglione R. Artimie cardiache rare dell’eta pediatria. Clin. Pediatr. (Phila.). 1963;45:656–683.
-
- Schwartz PJ, Priori SG, Dumaine R, Napolitano C, Antzelevitch C, Stramba-Badiale M, Richard TA, Berti MR, Bloise R. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med. 2000;343:262–7. [PubMed]
-
- Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, Horie M, Schulze-Bahr E, Denjoy I. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation.2006;113:783–90. [PubMed]
-
- Siem G, Früh A, Leren TP, Heimdal K, Teig E, Harris S. Jervell and Lange-Nielsen syndrome in Norwegian children: Aspects around cochlear implantation, hearing, and balance. Ear Hear.2008;29:261–9. [PubMed]
-
- Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336:1562–7. [PubMed]
-
- Thomas G, Killeen MJ, Gurung IS, Hakim P, Balasubramaniam R, Goddard CA, Grace AA, Huang CL. Mechanisms of ventricular arrhythmogenesis in mice following targeted disruption of KCNE1 modelling long QT syndrome 5. J Physiol. 2007;578:99–114. [PMC free article] [PubMed]
-
- Towbin JA, Wang Z, Li H. Genotype and severity of long QT syndrome. Drug Metab Dispos.2001;29:574–9. [PubMed]
-
- Tranebjaerg L. Jervell and Lange-Nielsen syndrome. In: Willems PJ, ed. Genetic Hearing Loss.New York, NY: Marcel Dekker Publications; 2004:117-34.
- Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M. Jervell and Lange-Nielsen syndrome: a Norwegian perspective. Am J Med Genet. 1999;89:137–46. [PubMed]
42. Tristani-Firouzi M, Jensen JL, Donaldson MR, et al., Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome) in J. Clin. Invest., vol. 110, nº 3, 2002, pp. 381–8, DOI:10.1172/JCI15183, PMC 151085,PMID 12163457.
-
- Tyson J, Tranebjaerg L, McEntagart M, Larsen LA, Christiansen M, Whiteford ML, Bathen J, Aslaksen B, Sorland SJ, Lund O, Pembrey ME, Malcolm S, Bitner-Glindzicz M. Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Hum Genet. 2000;107:499–503. [PubMed]
-
- Wang DW, Desai RR, Crotti L, Arnestad M, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Rognum T, Schwartz PJ, George AL. Cardiac sodium channel dysfunction in sudden infant death syndrome.Circulation. 2007;115:368–76. [PubMed]
-
- Wang RR, Li N, Zhang YH, Wang LL, Teng SY, Pu JL. Novel compound heterozygous mutations T2C and 1149insT in the KCNQ1 gene cause Jervell and Lange-Nielsen syndrome. Int J Mol Med.2011;28:41–6. [PubMed]
-
- Wang Z, Li H, Moss AJ, Robinson J, Zareba W, Knilans T, Bowles NE, Towbin JA. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome. Mol Genet Metab.2002;75:308–16. [PubMed]
-
- Winbo A, Stattin EL, Diamant UB, Persson J, Jensen SM, Rydberg A. Prevalence, mutation spectrum, and cardiac phenotype of the Jervell and Lange-Nielsen syndrome in Sweden. Europace.2012;14:1799–806. [PubMed]
-
- Winbo A, Sandström O, Palmqvist R, Rydberg A. Iron-deficiency anaemia, gastric hyperplasia, and elevated gastrin levels due to potassium channel dysfunction in the Jervell and Lange-Nielsen Syndrome. Cardiol Young. 2013;23:325–34. [PubMed]
-
- Yanmei F, Yagin W, Haibo S, Huiqun Z, Zhengnong C, Dongzhen Y, Shankai Y. Cochlear implantation in patients with Jervell and Lange_Nielsen syndrome, and a review of the literature. Int J Pediatr Otorhinolaryngol. 2008;72:1723–9. [PubMed]
-
- Zehelein J, Kathoefer S, Khalil M, Alter M, Thomas D, Brockmeier K, Ulmer HE, Katus HA, Koenen M. Skipping of exon 1 in the KCNQ1 gene causes Jervell and Lange-Nielsen Syndrome. J Biol Chem. 2006;281:35397–403. [PubMed]
- Zhang S, Yin K, Ren X, Wang P, Zhang S, Cheng L, Yang J, Liu JY, Liu M, Wang QK. Identification of a novel KCNQ1 mutation associated with both Jervell and Lange-Nielsen and Romano-Ward forms of long QT syndrome in a Chinese family. BMC Med Genet. 2008;9:24.[PMC free article] [PubMed]
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Didascalia : |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Trasmissione “X-Iinked”
TRASMISSIONE “X LINKED”
Sordità Legate Al Cromosoma XTrasmissione ereditaria recessiva legata al sesso “X-Linked”
Indice
Cos’è l’ereditarietà recessiva legata all’X?
Ereditarietà dominante legata all’X
Analisi del portatore e analisi in gravidanza
Elenco di ipoacusie a Trasmissione “X-Iinked”
Ipoacusia Autosomica recessiva (AR)
Ipoacusia Autosomica dominante (AD)
Ipoacusia a Trasmissione mitocondriale
LE IPOACUSIE CONGENITE
Mentre fino a qualche anno fasi riteneva che le cause genetiche rappresentassero un terzo dei casi di ipoacusia nel bambino, attualmente grazie allo sviluppo delle diagnosi molecolari è stato possibile ricollegare a una causa genetica la maggioranza dei casi sporadici di ipoacusia, in precedenza classificati come “causa ignota”12.
Oggi pertanto si stima che circa il 75% delle ipoacusie congenite sia riferibile a cause genetiche (sindromiche nel 30%) e che la restante parte sia da ricondurre a cause ambientali, soprattutto infettive.
Le Ipoacusie genetiche
Le ipoacusie genetiche sono, nella maggior parte dei casi, malattie monogeniche ossia dovute alla lesione di un singolo gene che causa generalmente una lesione Cocleare.
La lesione è rappresentata da mutazioni della sequenza di base del DNA con modalità di alterazione differenti (sostituzione/inserzione/delezione).
Le modalità di trasmissione di una ipoacusia genetica sono:
Sordità Legate Al Cromosoma X Trasmissione ereditaria recessiva legata al sesso “X-Linked” Legata al cromosoma X (1%)
Rappresentano il 2-3% delle forme non sindromiche. Data la localizzazione della mutazione sul cromosoma X, la malattia colpisce generalmente solo i maschi i quali, possedendo un solo cromosoma X, sono definiti emizigoti per la mutazione. Le loro madri, avendo un cromosoma X normale ed uno con la mutazione, sono definite portatrici sane della malattia. Le donne portatrici sane trasmettono la malattia al 50% dei figli maschi.
Una malattia monogenica si dice a trasmissione ereditaria recessiva legata al sesso quando il gene coinvolto è localizzato su un cromosoma sessuale (X o Y); se il gene si trova sul cromosoma X si parla di trasmissione ereditaria legata all’X (X-linked) e il carattere patologico si manifesta di solito solo in individui di sesso maschile in quanto portano il solo cromosoma X con l’allele mutato (emizigoti); esempi di malattie recessive legate all’X sono la distrofia muscolare di Duchenne, sindrome dell’X-fragile, emofilia A e B. Un individuo di sesso femminile avendo una sola copia alterata di un gene (eterozigote) generalmente non manifesta la malattia ed è indicato come “portatore sano”. Nella Fig.1 è rappresentata la situazione più frequente, cioè l’incontro di una femmina “portatrice sana” e un maschio normale; per un dato gene del cromosoma X l’allele normale è indicato con X, quello mutato con x. Una madre eterozigote (Xx) per una mutazione produce ovuli che presentano l’allele nomale X o l’allele mutato x. Il padre da origine a spermatozoi con l’allele X o con il cromosoma Y. Dall’incontro di una femmina “portatrice sana” (eterozigote) e un maschio normale, a seconda del tipo di gameti che si fondono al momento della fecondazione si verifica che:
· in media il 50% dei figli maschi risulterà affetto (xY) e potrà trasmettere l’allele mutato x a tutte le sue eventuali figlie
· in media il 50% delle figlie di madri eterozigoti, sarà eterozigote e avrà una probabilità del 50% di trasmettere il cromosoma X con l’allele mutato (x) ai figli.

Fig.1- Trasmissione ereditaria di una malattia genetica legata al cromosoma X (X-linked)
Le malattie X-linked raramente possono colpire anche le donne. Una patologia può manifestarsi:
· in figlie (omozigoti xx) di madre eterozigote per la mutazione (Xx) e padre affetto (xY), in quanto ereditano l’allele mutato x da entrambi i genitori;
· in femmine eterozigoti (Xx) in cui si è inattivato preferenzialmente il cromosoma X con l’allele normale;
· in femmine con monosomia del cromosoma X (sindrome di Turner) in cui è presente solo il cromosoma X con la mutazione.
Con questa modalità il gene responsabile della sordità è situato sul cromosoma X, anziché su uno dei cromosomi autosomici.La sordità si manifesta nei maschi, che sono dotati di un solo cromosoma X (fig. 35c).Si ritiene che ‘1-2% delle sordità genetiche siano trasmesse con questa modalità, X-linked o legata al sesso, sono disturbi ereditati attraverso i geni sul cromosoma X. Il gene in causa è situato sul cromosoma X. I maschi che posseggono solo un cromosoma X sono più sensibili ai sintomi clinici ipoacusia di una malattia X-linked, questi trasmetteranno l’X portatore della mutazione genetica alle loro figlie che però saranno udenti. Nelle donne, infatti, l’X portatore della mutazione genetica è “compensato” dal secondo X normale (ipoacusia recessiva legata all’X).
Raramente, come nella sindrome di Alport, l’ipoacusia è dominante legata al cro- mosoma X e le femmine sono allora anche colpite, ma in modo meno grave dei maschi. Figura 2 mostra un esempio in cui la madre è un vettore inalterata e il padre ha una copia normale del gene. Il figlio colpito ha ereditato il cromosoma X anomalo dalla madre. Nei disturbi X-linked, i padri affetti di solito passano il gene anomalo per le loro figlie, ma non per i loro figli. Più del 80% dei pazienti con sindrome di Alport ha una malattia legata al cromosoma X associata a perdita dell’udito neurosensoriale, malattie renali e anomalie oculari (Kruegel, Rubel, e Gross, 2013; Rheault, 2012).
|
|
|

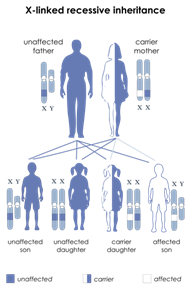
|
|
|

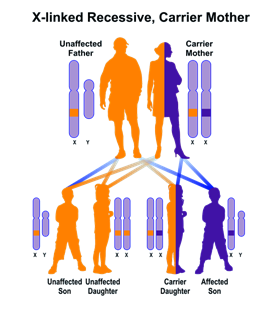
Fig. 2. ereditarietà X-linked
Legata al cromosoma Y (<1%)
Il gene in causa è situato sul cromosoma Y e pertanto interessa esclusivamente il sesso maschile. Non sono state riscontrate altre anomalie associate. Gli unici casi riscontrati appartengono ad una famiglia cinese con alterazione del gene POU3F4’.
Cos’è l’ereditarietà recessiva legata all’X?
Il cromosoma X ha molti geni che sono importanti per la crescita e lo sviluppo. Il cromosoma Y è più piccolo e ha meno geni. Le femmine hanno 2 cromosomi X (XX) e quindi se uno dei geni su un cromosoma X ha una mutazione, il gene normale sull’altro cromosoma X può compensare la copia mutata. Se questo accade la femmina è portatrice sana della malattia legata all’X. Essere portatori significa che non si ha la malattia ma si porta una copia del gene mutato. In alcuni casi le femmine mostrano segni leggeri della malattia. I maschi hanno un cromosoma X e un Y (XY) e quindi se uno dei geni su un cromosoma X del maschio ha una mutazione, non c’è un’altra copia di quel gene per compensare la copia mutata. Questo significa che sarà affetto dalla malattia, che quando viene ereditata in questo modo viene chiamata malattia recessiva legate all’X. Alcuni esempi di malattie legate all’X includono la Sindrome di Norrie , la Sindrome di Alport,sindrome oto-palato-digitale (OPD) l’emofilia e la distrofia muscolare Duchenne.

Schema di trasmissione genetica con gene X recessivo da madre portatrice sana
Fig.3: Come vengono trasmesse le malattie legate all’X dalle femmine portatrici
Nel caso si tratti di anomalie recessive , la malattia colpisce praticamente solo i maschi nati da madri portatrici (clinicamente sane).

Ereditarietà dominante legata all’X
Sebbene la maggior parte di malattie legate al cromosoma X segua una trasmissione recessiva, vi sono rare forme di malattie genetiche legate al cromosoma X che seguono una trasmissione dominante. Questo vuol dire che anche se una femmina eredita un cromosoma X normale ed un cromosoma X con una mutazione, il gene mutato ha il sopravvento sul gene normale e quindi si manifesta la malatia. Se un maschio eredita il cromosoma X con la mutazione sarà affetto perchè i maschi hanno un solo cromosoma X. Una femmina ha quindi 1 probabilità su 2 (50%) di trasmettere la malattia sia ai figli maschi che alle femmine. Un maschio affetto avrà tutte le figlie femmine affette ma tutti i maschi normali (perchè a questi trasmetterà solo il cromosoma Y).
Le malattie X-linked dominanti sono rare. In questo caso una madre affetta ha un 50% di probabilità d trasmettere la malattia ai propri figli (maschi o femmine che siano), mentre un padre affetto trasmette la malattia solo alle figlie.
Punti da ricordare
· Le femmine portatrici hanno il 50% di possibilità di passare il gene mutato. Se un figlio eredita un gene mutato da sua madre, allora sarà affetto dalla malattia. Se una figlia eredita un gene mutato sarà portatrice come sua madre.
· Un maschio che ha la malattia legata all’X passerà sempre il gene mutato a sua figlia che sarà quindi una portatrice. Un maschio non passerà mai un gene mutato a suo figlio.
· Un gene mutato è presente per tutta la vita e raramente può essere corretto.
· Un gene mutato non è qualcosa che può essere preso da altre persone. Quindi si può anche essere donatore di sangue, per esempio (ad eccezione di soggetti con malattie genetiche del sangue).
· Le persone spesso si sentono colpevoli a causa di una malattia genetica che si trova in famiglia. È importante ricordare che non è colpa di nessuno e nessuno ha fatto nulla per causarne la comparsa.
Se una femmina portatrice ha una figlia, passerà o il cromosoma X con il gene normale, o il cromosoma X con il gene mutato. Ogni figlia quindi 1 probabilità su 2 (il 50%) di ereditare il gene mutato. Se questo accade la figlia sarà portatrice come la madre. C’è anche 1 probabilità su 2 (il 50%) che la figlia erediti il gene normale. Se questo accade sarà totalmente sana rispetto la malattia. Questo rischio rimane lo stesso per ogni figlia.
Se un maschio che ha la malattia legata all’X ha una figlia le trasmetterà sempre il gene mutato. Questo perché i maschi hanno solamente un cromosoma X e passano sempre questo alle loro figlie. Tutte le sue figlie saranno portatrici. Le figlie di solito non avranno la malattia ma sono a rischio di avere figli affetti.
Se un maschio che ha una malattia legata all’X ha un figlio, suo figlio non erediterà mai il gene mutato sul cromosoma X. Questo perché i maschi sempre passano il loro cromosoma Y ai loro figli ( se passano il loro cromosoma X avranno una figlia).
Cosa accade se un bambino é la prima persona nella famiglia ad avere la malattia?
Qualche volta un bambino nato con una malattia genetica legata all’X può essere la prima persona ad essere affetta nella famiglia. Questo può accadere perché una nuova mutazione genetica é avvenuta per la prima volta o nell’uovo o nello spermatozoo che originarono il bambino. Quando questo accade, nessuno dei genitori del bambino é portatore. Comunque il bambino affetto che ora ha il gene mutato, può passarlo ai suoi bambini.
Analisi del portatore e analisi in gravidanza
Diverse opzioni possono essere disponibili per le persone che hanno una storia familiare di malattia genetica legata all’X. L’analisi del portatore può essere disponibile per le femmine per vedere se sono portatrici del gene mutato. Questa informazione può essere utile quando si pianificano gravidanze. Per alcune malattie legate 8 all’X é possibile fare l’analisi in gravidanza per vedere se il bambino ha ereditato la malattia (ulteriori informazioni circa queste analisi sono disponibili negli opuscoli CV e amniocentesi). In questi casi si consiglia di discuterne con lo specialista in Genetica Medica (consulenza genetica).
Situazioni riguardanti altri membri familiari Se qualcuno in famiglia ha una malattia legata all’X o é portatore, potrebbe essere utile discutere con altri membri della famiglia. In questo modo si offre ad altre femmine membri della famiglia l’opportunità di fare un’analisi del sangue per vedere se sono portatrici, se lo desiderano. Queste informazioni possono essere utili ad aiutare altri membri della famiglia a fare una diagnosi. Questo potrebbe essere particolarmente importante per i membri della famiglia che hanno già figli o che vorrebbero avere figli in futuro. Alcune persone trovano difficile di parlare della malattia genetica ad altri membri della famiglia. Essi possono preoccuparsi di causare ansia nella famiglia. In alcune famiglie le persone hanno perso il contatto con i parenti e può essere difficile contattarli. Lo specialista in genetica ha molta esperienza con le famiglie in queste situazioni ed è in grado di offrire un aiuto per discutere la situazione con altri membri della famiglia
Elenco di ipoacusie a Trasmissione “X-Iinked” Legata al cromosoma X
La sindrome di Alport è una condizione genetica caratterizzata dalla progressiva perdita di funzione renale e uditiva. La sindrome di Alport può inoltre interessare gli occhi. La presenza di sangue nelle urine (ematuria) è quasi sempre riscontrabile nella patologia.Nella maggior parte (80-85%) dei pazienti affetti da Sindrome di Alport, la patologia è trasmessa con un’ereditabilità legata all’X, dovuta a mutazioni nel gene COL4A5. Una condizione si considera legata all’X se il gene coinvolto nella malattia è localizzato sul cromosoma X. Nei soggetti di sesso maschile, che possiedono un solo cromosoma X, una copia alterata del gene COL4A5 è sufficiente per causare la Sindrome di Alport in forma severa, fino ad arrivare all’insufficienza renale. Nelle femmine, che possiedono due cromosomi X, la mutazione di una copia di COL4A5 solitamente provoca ematuria, ma nella maggior parte dei casi non arrivano mai all’insufficienza renale. Una interessante caratteristica delle malattia a trasmissione legata all’X è che il padre non le trasmette mai ai figli maschi.
La sindrome di Norrie (o malattia di Norrie, o displasia oculo-acustico-cerebrale), è una malattia genetica molto rara, legata al generecessivo NDP presente sul cromosoma X, che causa cecità, sordità e ritardo mentale. Colpisce solo i maschi, mentre le femmine sono portatrici sane.
La sindrome oto-palato-digitale (OPD) è una malattia genetica rara, che associa displasia scheletrica, ipoacusia, palatoschisi e viso caratteristico (con ipertelorismo, radice nasale allargata, bozze frontali prominenti, naso piccolo piatto e rime palpebrali rivolte in basso e verso l’esterno). La patologia riconosce un’eziologia genetica ed una modalità di trasmissione di tipo X-linked dominante (tipo I) o recessiva (tipo II). Il gene della forma tipo II responsabile è stato mappato al locus Xq28.
La sindrome di Wildervanck, nota anche come sindrome cervicooculoacoustica, è una malattia genetica rara che colpisce soprattutto le donne. La malattia è caratterizzata da sordità congenita percettiva, una condizione scheletrica nota come sindrome di Klippel-Feil (KFS); anomalie di alcuni movimenti dell’occhio con Retractio bulbi (ad esempio, la sindrome di Duane); Nella maggior parte dei casi, la sindrome Wildervanck sembra verificarsi in modo casuale per ragioni sconosciute (sporadicamente). Poiché la malattia colpisce soprattutto le donne, alcuni ricercatori suggeriscono che la sindrome Wildervanck può essere trasmessa come carattere dominante X-linked.
altre ipoacusie X-Linked sono ad esampio:DFNX1 (DFN2),DFNX2 (DFN3), DFNX4 (DFN6)
DFNX (per Deafness X Linked) (per Deafness Autosomal Dominant), DFNB (per Deafness Autosomal Recessive), Si attribuisce poi un numero per ordine di scoperta (Tabelle I—IV).
Tabella I – Lista dei loci e dei geni delle ipoacusie X-Linked (da Van Camp e Smith 2010)
|
Locus |
Posizione |
Gene |
Referenze |
|
DFNXI |
Xq22 |
PRPSI |
Liuetal.. 2010 |
|
DFNX2 |
Xg21.1 |
POU3F4 |
DeKoketal., 1995 |
|
DFNX3 |
Xp21.2 |
sconosciuto |
Lalwani et al., 1994 |
|
DFNX4 |
Xp22 |
sconosciuto |
del Castillo et aI.. 1996 |
|
DFNX5 |
Xq23-q27.3 |
sconosciuto |
Wang et al , 2006 |
|
Locus (OMIM) |
Gene (OMIM) |
Riferimento |
|
DFNX1 (DFN2) |
||
|
DFNX2 (DFN3) |
||
|
DFNX4 (DFN6) |
||
Ipoacusia Autosomica recessiva (AR) (76%)
E’ la modalità più frequente. infatti si stima che circa tre quarti delle ipoacusie non sindromiche si trasmettano in modalità AR.Quando una malattia si trasmette in ipoacusia a trasmissione autosomica recessiva della moda, due copie del gene anomalo causano la malattia. Gli individui che ereditano un solo gene anomalo e un gene normale sono indicati come vettori e non sono colpiti dalla malattia. I genitori portatori di un solo allele anomalo del gene in causa (portatori eterozigoti) Sono normoudenti e spesso sono inconsapevoli del loro stato di portatore fino a quando non hanno un figlio affetto. Gli individui affetti da una malattia autosomica recessiva di solito sono il risultato di accoppiamenti tra due vettori. La Fig.1 illustra il potenziale risultati di tale accoppiamento, in cui il 25% della prole sono affetti da disturbo in questione, il 50% sono portatori sani, e il 25% sono liberi della malattia e con il gene anormale. La Consanguineità , o la presenza di un comune antenato tra la coppia, aumenta il rischio di una malattia autosomica recessiva. La Sindrome di Usher mostra un modello di ipoacusia autosomica recessiva. La prevalenza della sindrome di Usher è di 3 a 4 soggetti affetti si 100.000 nati (Ahmed, Riazuddin, Riazuddin, & Wilcox, 2003), con tassi di prevalenza più elevati in alcune popolazioni, tra cui la popolazione Acadian in Louisiana (Ouyang et al., 2003) e la popolazione ebrea Ashkenazi (Guha et al., 2012). Il tipo più grave di sindrome di Usher è caratterizzata da una sordità profonda neurosensoriale congenita e da progressiva perdita della vista (Liu et al., 2013). Garantire una diagnosi precoce della sindrome di Usher può aiutare i pazienti a prepararsi per il loro futuro. La sindrome di Usher può rappresentare la metà di tutti i casi di concomitante sordità / cecità (Yan e Liu, 2010).

Fig.1 ereditarietà autosomica recessiva

Ipoacusia Autosomica dominante (AD) (22%)
In un modello di ereditarietà autosomica dominante , un bambino eredita una copia normale di un gene da un genitore e un gene anomalo dall’altro genitore. Nelle famiglie affette da sordità autosomica dominante, uno dei genitori è sordo e porta su un singolo allele del gene la mutazione patogena, che trasmetterà a metà dei suoi figli, che quindi saranno sordi. Il gene anomalo domina il gene normale, così una copia di un gene anormale è sufficiente a causare una malattia autosomica dominante (Fig. 2).La mutazione dell’allele interessato può. per esempio. produrre una proteina anormale, che impedisce la funzione della proteina normale prodotta dall’allele sano. La Sindrome di Waardenburg è la causa più comune di perdita di udito sindromica autosomica dominante, che colpisce 1 su 42.000 persone (Read & Newton, 1997). Quando un genitore ha la sindrome di Waardenburg, ogni bambino di quel genitore ha una probabilità del 50% di essere affetti da sindrome di Waardenburg, assumendo l’altro genitore non ha la sindrome. Il restante 50% della prole in questo accoppiamento sarà inalterato. Caratteristiche tipiche della sindrome di Waardenburg includono perdita dell’udito neurosensoriale, un ciuffo bianco, occhi celesti o di colore diverso, e gli occhi distanziati. Una grande quantità di espressività variabile esiste in Waardenburg sindrome, in modo che i pazienti possono avere una qualsiasi combinazione di caratteristiche, tra cui un udito normale (de Sousa Andrade et al., 2012). Waardenburg sindrome rappresenta circa il 2% dei casi di sordità profonda congenita (de Sousa Andrade et al., 2012).Va notato che, in rari casi, alcuni disturbi autosomica dominante verificarsi a causa di una nuova mutazione nel bambino e nessuno dei genitori ha il disturbo. L’espressività è però variabile in questa modalità di trasmissione: la gravità della ipoacusia può essere variabile tra i vari soggetti colpiti.
Quando l’ipoacusia è sindromica i segni associati alla sindrome possono essere assenti o lievi in alcuni sordi della famiglia e alcuni membri portatori dell’ alterazione genetica possono essere udenti.


Fig.2 ereditarietà autosomica dominante
Ipoacusia a Trasmissione mitocondriale (1%)
Il genoma mitocondriale è un frammento di DNA situato nel mitocondrio. La trasmissione dell’anomalia mitocondriale è esclusivamente matrilineare. Quando un gene dell’ipoacusia è situato sul DNA mitocondriale. uomini e donne possono essere sordi, ma solamente le donne potranno trasmettere l’ipoacusia ai loro figli che saranno in teoria tutti sordi.
Sebbene mutazioni mitocondriali geni rappresentano una piccola percentuale di casi di perdita di udito non sindromica, sono importante notare per la loro modalità di trasmissione e la loro associazione con una insolitamente alta suscettibilità alla ototossicità da aminoglicosidici (Chen, egli, Fu, e Dong, 2011). I mitocondri sono organelli cellulari responsabili della produzione di energia. Si trovano al di fuori del nucleo e hanno il loro DNA e geni, separati dai geni nucleari. I mitocondri sono presenti in cellule uovo, ma non in cellule spermatiche, quindi sono ereditate esclusivamente attraverso la linea materna. Una donna con una mutazione mitocondriale passerà la mutazione a tutti i suoi figli, mentre un uomo con una mutazione mitocondriale non dia mai a nessuno dei suoi figli. Eredità mitocondriale è dimostrato in Figura 4. Le mutazioni nel mitocondriale MT-RNR1 gene causa un aumento della suscettibilità alle ototossicità aminoglicoside indotta, a volte causando sordità profonda dopo una sola dose di antibiotici aminoglicosidici (Van Camp & Smith, 2000). Lo screening neonatale per le mutazioni mitocondriali che portano a ototossicità aminoglicoside indotta non è ancora disponibile. Il test genetico dovrebbe essere richiesto, se un modello di ereditarietà materna è osservato (Figura 4) e il bambino è influenzata dalla somministrazione di antibiotici aminoglicosidici. I test genetici di una mutazione mitocondriale può identificare altri membri della famiglia o future gravidanze a rischio di insorgenza precoce perdita sostanziale dell’udito, quindi consentire l’adozione di misure preventive. In questi casi, i genitori si consiglia di evitare la somministrazione di antibiotici aminoglicosidi e cercare trattamenti antibiotici alternativi.
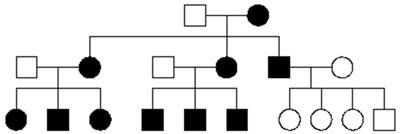
Figura 4. ereditarietà mitocondriale
L’ipoacusia è spesso severa e si manifesta dopo esposizione a antibiotici aminoglicosidici. Alcune fonne sono sindromiche e coinvolgono in particolare il sistema nervoso:
MELAS: Mitochondrial Encephalopathy Lactic Acidosis Stroke-like episodes
KSS: Kearns — Sayre Syndrome
MERRF: Myoclonic Epilepsy and Ragged Red Fibers
MDD: Maternal Inherited Diabetes and Deafness.
Più di 60 geni implicati nella perdita di udito non sindromica sono stati identificati (Van Camp & Smith, 2013). Circa l’80% dei casi di perdita di udito non sindromica genetiche sono ereditate in modo autosomico recessivo, mentre quasi il 20% dimostra una modalità autosomica dominante di eredità (Hilgert, Smith, e Van Camp, 2009). X-linked e geni mitocondriali rappresentano una piccola percentuale di casi (Hilgert et al., 2009). Il gene più comune che causa la perdita di udito non sindromica è il GJB2gene, noto anche come Connexin 26 o DFNB1 (Angeli, Lin, e Liu, 2012). I conti gene connessina 26 per circa la metà di tutte le cause autosomica recessiva di sordità sindromica (Kenneson, Van Naarden Braun, e Boyle, 2002). La maggior parte di questi pazienti sono nati da genitori normali, udito, che sono entrambi portatori di mutazioni nello stesso gene (Angeli et al., 2012).
Loci e geni coinvolti nelle ipoacusie non sindromiche
L’analisi molecolare in famiglie con casi di ipoacusia genetica ha permesso di definire il locus ossia le regioni del genoma contenenti il gene interessato. Il gene spesso è stato identificato in un secondo tempo e in qualche caso un locus ha dimostrato di contenere i due geni dell’ipoacusia. È stata fissata una codifica internazionale pci denominare ogni locus delle ipoacusie non sindromiche. Il codice inizia con DFNA
Tabella II a – Lista dei loci e dei geni delle ipoacusie non sindromiche dominanti
(daVan Camp e Smith 2010
|
Locus |
Posizione |
Gene Referenze |
|
DFNAI |
5q31 |
DIAPHI Léon et al, 1992 |
|
Lynchetal., 1997 |
||
|
DFNA2A |
lp34 |
KCNQ4 Coucke et al., 1994 |
|
DFNA2B |
lp35 I |
GJB3(in Xiaetal., 1999 passato CX3I) |
|
DFNA3A |
13q1 1-q12 |
GJB2(in Chaib et al., 1994 |
|
DFNA3B |
13q12 |
GJB6(in Grifaetal,. 1999 passato CX3O) |
|
DFNA4 |
19q13 |
MYH14 Chen et al., 1995, Donaudy et al, 2004 |
|
DFNA5 |
7p15 |
DFNA5 Van Camp et al, 1995 |
|
DFNA6 |
4pl6.3 |
WFSI Lesperance et al., 1995: Van Camp et al., 1999; |
|
DFNA7 |
1q21-q23 |
sconosciuto Fagerheim et al.. 1996 |
|
DFNA8 |
vedi DFNAI2 |
|
|
DFNA9 |
14ql2-q13 |
COCH Manolis et al.. 1996 |
|
DFNÀIO |
6q22-q23 |
EYA4 ONeili et aI. 1996 |
|
Wayne et al.. 2001 |
||
|
DFNAI I |
1 lql2 3-q2l |
MYO7A Tamagawa et al.. 1996 |
|
DFNA12 |
I Iq2224 |
TECTA Verhoeven et aI.. 1997 |
|
Verhoeven et al,. 1998 |
||
|
DFNAI3 |
6p2l |
COLI 1A2 Brown et al. 1997 |
|
McGuirt et al.. 1999 |
||
|
DFNAI4 |
vedi DFNA6 |
|
|
DFNAI5 |
5g3I |
POIJ4F3 Vahava et al.. 1998 |
|
DFNAI6 |
2q24 |
sconosciuto Fukushima et al., 1999 |
|
DFNAI7 |
22q |
MYH9 Lalwaniet al. 1999 |
|
DFNAI8 |
3q22 |
Sconosciuto Bonsch et al.. 200! |
|
DFNAI9 |
10 (pericentr.) |
sconosciuto The Molecular Biology of Hearing and Deafness, Bethesda, October 8l I. 1998 (Green et al., abstract 107) |
|
DFNA2O |
17q25 |
ACTG1 Moreli et al .2000. Yang et al,. 2000, Zhu et aL. 2003. van |
|
DFNA2I |
6p21 |
sconosciuto Kunst et al.. 2000 |
|
DFNA22 |
6gl3 |
MYO6 Melchionda et al.. 2001 |
|
DFNA23 |
I4q2l-q22 |
sconosciuto Salam et al.. 2000 |
|
DFNÀ24 |
4q |
sconosciuto Hafner et al.. 2000 |
|
DFNA25 |
12g21-24 |
sconosciuto Greene et aI.. 1999 |
|
DFNA26 |
vedi DFNA2O |
|
|
DFNA27 |
4q12 |
sconosciuto Frideil et aI.. 999 |
|
DFNA2S |
8q22 |
TFCP2L3 Anderson et al.. 1999 Peters et aI.. 2002 |
|
DFNA29 |
||
|
DFNA3() |
I 5q5-26 |
sconosciuto Mangino et al 200! |
|
DFNA3I |
6p2i.3 |
Sconosciuto |
Snoeckx et ai. 2004 |
|
DFNA32 |
i1p15 |
sconosciuto |
Li et ai.. 2000 |
|
DFNA33 |
13g34-gter |
sconosciuto |
Bonsch et ai.. 2009 |
|
DFA34 |
1g44 |
Kurinìa et ai., 2000 |
|
|
DFNA35 |
|||
|
DFNA36 |
9g13-g2i |
TMCI |
Kurimaetai.,2002 |
|
DFNA37 |
1p21 |
Taiebizadeh Ct ai.. 2000 |
|
|
DFA38 |
vedi DFNA6 |
Xiaoetai.. 2001 |
|
|
DFNA39 |
4g21.3 |
DSPP |
|
|
DFA40 |
i6pi2 |
||
|
DFNA4I |
12g24-gter |
sconosciuto |
Bianton_et_ai.,_2002 |
|
DFNA42 |
5g3 1.1 -g32 |
sconosciuto |
Xia et ai., 2002 |
|
DFNA43 |
2pi2 |
Fiex et ai,, 2003 |
|
|
DFA44 |
3q2829 |
CCDC5O |
Modarnio-Hoybjor et ai., 2003; Modamio-Hoybjor et ai. 2007 |
|
DFNA45 |
|||
|
DFNA46 |
|||
|
DFNA47 |
9p2i-22 |
sconosciuto |
D’Adamo et ai., 2003 |
|
DFNA$8 |
i2gi3-gi4 |
MYOiA |
D’Adamo et ai.. 2003. Donaudy et ai.. 2003 |
|
DFNA49 |
ig2i-g23 |
sconosciuto |
Moreno-Peiayo et ai.. 2003 |
|
DFNA50 |
7g32.2 |
MIRN96 |
Modamio-Hoybjor et ai.. 2004: Mencia et ai.. 2009 |
|
DFA5I |
9g2i |
TJP2 |
Waishetai.. 2010 |
|
DFNA52 |
4g28 |
||
|
DFN53 |
I 4g Il .2-g 12 |
sconosciuto |
Yan CI ai.. 2005 |
|
DFA54 |
5g3 I |
sconosciuto |
Gurtier et ai., 2004 |
|
DF>A55 |
|||
|
DFA56 |
|||
|
DFNA57 |
I9pI3.2 |
sconosciuto |
Bonsch et aI., 2008 |
|
DFNA5 |
2pi2-p2I |
sconosciuto |
Lezirovitz et ai.. 2009 |
|
DFNA59 |
i ipI4.2gi2.3 |
sconosciuto |
Chatterjee cI ai.. 2009 |
|
DFNA6(I |
2g2i.3-g24.i |
Liu XZet ai. ARO meeting. Denver. February 2007. |
|
Tabella III – Lista dei loci e dei geni delle ipoacusie non sindromiche recessive (da Van Camp e Smith 2010)
|
Locus |
Posizione Gene |
Referenze |
|
DFNB1A |
13q1 1-12 GJB2(in passato CX26) |
Guilford et al, 1994 |
|
Keiscll et al., 1997 |
||
|
DFNBIB |
13g12 GJB6(in passato CX3O) |
Del Castillo et aI., 2002 |
|
DFNB2 |
11q13.5 MYO7A |
— |
|
DFNB3 |
17pl 1.2 MYOI5A |
Fricdman et al., 1995 Wang et al., 1998 |
|
DFNB4 |
7q31 SLC26A4 |
Baldwin et al. 1995 Li Ct al., 1998 |
|
DFNB5 |
l4q12 sconosciuto |
Fukushima et al., 1995 |
|
(Vedi Nota 1) |
||
|
DFNB6 |
3p14—p21 TMIE |
Fukushima et al, 1995 Naz et al, 2002 |
|
DFNB7/1 I |
9g13-g21 TMCI |
Jain ci al.. 1995, Scoti ci al., l996,Kurima ci al.. 2002 |
|
DFNB8/DFNBIO |
21q22 TMPRSS3 |
Veskeetal., 1996, |
|
DFNB9 |
2p22-p23 OTOF |
Chaib ci al. 1996 |
|
(Vedi Nota 2) |
Yasunaga et al.. 1999 |
|
|
DFNBIO |
vedi |
|
|
DFNBI I |
vedi DFNB7 |
|
|
DFNBI2 |
10q21-q22 CDH23 |
Chaib ci al. 1996 Borketal.. 2001 |
|
DFNB 13 |
7g34-36sconosciuto |
Mustapha et al.. 1998 |
|
DFNBI4 |
7g31 sconosciuto |
Mustapha ci al.. 1998 |
|
DFNBI5 |
3q2l-q25 sconosciuto l9p13 |
Chen ci al, 1997 |
|
DFNB16 |
15g21-q22 STRC |
Campbcll ci al. 1997. Verpv ct al., 2001 |
|
DFNB17 |
7g31 Sconosciuto |
Greinwald ci al.. 1998 |
|
DFNB18 |
1 lpl4-l5.l USHIC |
Jain ci al, 1998. Ouyang ci aI., 2002; Ahmed ci al.. 2002 |
|
DFNB19 |
iSpi 1 sconosciuto |
The Molecular Biology of Hearing and Dcafness meeting Bcthcsda. October 8-11. 1998 (Green et al., abstract 108) |
|
DFNB2O |
1 lg25-gicr sconosciuto |
Moynihan ci al., 1999 |
|
DFNB2I |
I lg TECTA |
Mustapha ci al.. 1999 |
|
DFNB22 |
l6pl2.2 OTOA |
Zwaencpoel ci al.. 2002 |
|
DFNB23 |
lOpI l.2-q21 PCDI-115 |
Ahmed ci al. 2003 |
|
DFNB24 |
I lg23 RDX |
Khan et al.. 2007 |
|
DFNB25 |
4pl3 GRXCRI |
Schraders ci al.. 2010 |
|
DFNB26 |
4q3l sconosciuto |
Riazuddin Ct al,. 2000 |
|
(Nota 3) |
||
|
DFNB27 |
2g23-g3 I sconosciuto |
Pulleyn ci al.. 2000 |
|
DFNB28 |
22q13 TRIOBP |
Walsh ci al . 2000 |
|
DFNB29 |
21g22 CLDNI4 |
Wilcox ci al.. 2001 |
|
DFNB3O |
lOpI 1.1 MYO3A |
Walsh ci aI.. 2002 |
|
DFNB3 I |
9q32-q34 Wl-IRN |
Mustapha ci al . 2002 |
|
Mburu et al., 2003 |
||
|
DFNB32 |
lpl3.3-22.l GPSM2 |
Masmoudi et al., 2003; Walsh et al., 2010 |
|
DFNB33 |
9g34.3 sconosciuto |
Medlej-Hashim et aI., 2002 |
|
DFNB34 |
||
|
DFNB35 |
14q24.1- ESRRB |
Ansaret al., 2003: Collin et al,, 2008 |
|
DFNB36 |
I p36.3 ESPN |
Naz et al.. 2004 |
|
DFNB37 |
6g13 MYO6 |
Ahmedetal., 2003 |
|
DFNB38 |
6g26-g27 Sconosciuto |
Ansaret aL, 2003 |
|
DFNB39 |
7g21.l HGF |
Schultz etal., 2009 |
|
DFNB4O |
22g Sconosciuto |
Delmaghani et aI.. 2003 |
|
DFNB41 |
||
|
DFNB42 |
3q13.31- sconosciuto |
Aslam et aL, 2005 |
|
DFNB43 |
||
|
DFNB44 |
7pl4, 1- Sconosciuto |
Ansar et al., 2004 |
|
DFNB45 |
1g43-Q44 sconosciuto |
Bhatti et al., 2008 |
|
DFNB46 |
l8pll.32- sconosciuto plI.31 |
Miretal., 2005 |
|
DFNB47 |
2p2S,l- sconosciuto |
Hassan et al., 2005 |
|
DFNB48 |
15q23-g25.l sconosciuto |
Ahmad et al.. 2005 |
|
DFNB49 |
5q12 3- MARVELD2 |
Rainzan et al ,2004: Riazuddin et al ,2006 |
|
DFNB5O |
12g23 sconosciuto |
|
|
DF\B5I |
I 1p13-p12 sconosciuto |
Shaikh et al., 2005 |
|
DFNB52 |
||
|
DFNB53 |
6p2l.3 COLI 1A2 |
Chen et al., 2005 |
|
DFNB54 |
||
|
DFNB55 |
4g12-g13.2 sconosciuto |
Irshad et al., 2005 |
|
DFNB56 |
||
|
DFNB57 |
10q23.1- sconosciuto |
|
|
DFNB5S |
2q14 I- sconosciuto |
R Smith, inedito |
|
g21.2 |
||
|
DFNB59 |
2q3l 1- PJVK |
Delmaghani et al .2006 |
|
g31.3 |
||
|
DFNB6O |
5g22-g3 I Sconosciuto |
R. Smith. inedito |
|
DFNB6I |
7g22.I SLC26A5 |
Liu et al.. 2003 |
|
DFNB62 |
12pl3 2- sconosciuto |
Ali et al,, 2006 |
|
p1123 |
||
|
DFNB63 |
I Iq13.2- LRTOMT! COMT2 |
Du et al.. 2008: Ahmed et al., 2008 |
|
g13.4 |
||
|
DFNB6.i |
||
|
DFNB65 |
20q13 2- sconosciuto |
Tariq et al . 2006 |
|
g13.32 |
||
|
DFNB66 67 |
6p2l.2-22.3 LHFPL5 |
TIiIi Ct aI. 2005. Shabbiret al.. 2006: Kalay et al., 2006 |
|
DFB6 |
Vedi |
|
|
DFNB66 |
||
|
DFNB6S |
l9pl3.2 sconosciuto |
Santos et al.. 2006 |
|
DFNB69 |
Nota 1. DFNB5 è stata segnalata originariamente come DFNB4.
Nota 2. DFNB9 è stato segnalato in origine come DFNB6.
Nota 3. DFNB26 è soppresso da modificatore dominante DFNM1.
Esistono molti tipi di test genetici disponibili che colpiscono diverse porzioni del genoma. I disordini genetici possono essere causati da anomalie nelle varie regioni; il metodo per identificare il difetto genetico dipende dal tipo di sindrome genetica o anormalità sospettata e, quindi, la loro nota difetto genomico. Da larga scala per piccoli cromosoma o DNA modifiche, disturbi genetici possono derivare da una variazione del numero di cromosomi, delezione o l’aggiunta di una porzione di una regione cromosomica o una mutazione del DNA all’interno di un singolo gene.
Citogenetica (cariotipo o cromosoma) test viene utilizzato per rilevare anomalie cromosomiche. Le variazioni del numero di cromosomi in ogni cellula sono spesso incompatibili con la vita, perché ogni cromosoma contiene migliaia di geni. Perdita o guadagno di un numero così elevato di geni è molto dannoso per lo sviluppo. La forma più comune di sopravvivere cromosoma numero aberrante è la sindrome di Down, che colpisce 1 su 700 nascite (Ramia, Musharrafieh, Khaddage, e Sabri, 2013). La sindrome di Down, detta anche Trisomia 21 , i risultati di una copia in più del cromosoma 21. Gli individui con sindrome di Down, quindi, hanno un numero di cromosomi di 47 invece del solito 46. sindrome di Down è una delle principali cause di disabilità intellettiva e deficit dell’udito sono riportati nel 34% al 78% dei casi pediatrici (Raut et al, 2011;. Shott, Joseph, e Heithaus, 2001). La perdita dell’udito può essere conduttiva, neurosensoriale, o misto (Austeng et al., 2013), e l’otite media è molto comune (Ramia et al., 2013). La maggior parte dei casi di sindrome di Down sono sporadici, ma un piccolo numero di casi sono ereditate (Verma, Lall, e Dua Puri, 2012). L’età avanzata materna è un importante fattore di rischio (Smith & Visootsak, 2013). La diagnosi viene effettuata mediante analisi cromosomica di sangue o, nel periodo prenatale, per l’analisi cromosomica delle amniotico prelievo dei villi coriali o liquido. I campioni sono esaminati al microscopio per analizzare il numero totale di cromosomi presenti e per determinare quali cromosomi sono duplicati o mancanti. Le cellule vengono esposte e cromosomi sono disposti in un cariotipo (vedi Figura 5). Test citogenetica è estremamente utile nella diagnosi di trisomie e altre anomalie cromosomiche. Tuttavia, riarrangiamenti genetici più piccole potrebbero non essere visibili a causa della sua limitata risoluzione.
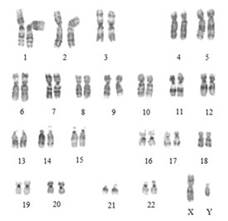
Figura 5. cariotipo maschile normale
I cromosomi con delezioni o duplicazioni troppo piccoli per essere visualizzati con i test citogenetici di routine può essere ulteriormente analizzate da FISH (a fluorescenza in situ ibridizzazione). test FISH comporta l’applicazione di un breve segmento di DNA (sonda) che codifica per il disturbo sospetto marcato con un tag fluorescenti . La sonda a fluorescenza si collega alla sua sequenza complementare sul campione del paziente. Le cellule sono poi analizzati per la presenza o l’assenza del segnale di sonda fluorescente al microscopio. Il vantaggio di FISH è che è molto specifico per la diagnosi di microdeletions e microduplicazioni. Tuttavia, un disturbo particolare deve essere sospettata in modo che venga utilizzato la sonda FISH corretta. Un elenco di sindromi microdelezione si possono trovare sul sito web Springer Protocols (Schwartz & Graf, 2002). Un esempio di un saggio FISH è mostrata in Figura 6.

Figura 6. La diagnosi di sindrome di DiGeorge utilizzando la fluorescenza in situ ibridazione (FISH).
Segnale verde: normale controllo. Segnale Rosso: sindrome di DiGeorge regione del gene. Del (22): la cancellazione positiva del gene sul cromosoma 22 DiGeorge.
Negli ultimi anni, la tecnologia microarray è stata sviluppata per test genetici. Microarray ibridazione genomica comparativa (CGH) confronta il DNA dal paziente con DNA da un controllo normale. I campioni di DNA del paziente e di controllo sono etichettati con sonde fluorescenti di colore diverso e lasciati a competere per il legame alle migliaia di segmenti di DNA su un vetro “chip”. Questo viene poi analizzato da un laser e le differenze tra il DNA paziente e normale DNA di controllo vengono analizzati da un software specializzato. Gli utili e le perdite di DNA possono essere visualizzati da questa tecnologia, che offre un ampio panorama dell’intero genoma; è, quindi, non è necessario conoscere l’anomalia genetica esatto in cui si richiede questo test. Microarrays possono essere personalizzati per la ricerca di un particolare gruppo di disturbi. Ad esempio, un chip con segmenti di molti geni differenti associati con perdita di udito può essere creato per testare più geni sordità contemporaneamente. Una limitazione di microarray è risultato insolito che possono essere difficili da interpretare e le differenze tra una variante genetica normale e una vera patologia genetica (Evangelidou et al., 2013).

Figura 7. microarray Comparative Genomic Hybridization (CGH)
Alcuni tipi di perdita dell’udito sono causate da mutazioni o cambiamenti nel DNA che sono troppo piccole per essere rilevate dai metodi precedentemente menzionati. La mutazione analisi può essere effettuata da PCR (polymerase chain reaction ). PCR è una tecnica di amplificazione di un piccolo segmento di DNA per produrre milioni di copie. Questa tecnica genera un ampio campione di DNA che può essere analizzato visualizzando il campione attraverso elettroforesi su gel. Il cambiamento genetico più piccolo comporta la sostituzione di una coppia di basi di DNA con un altro, definita una mutazione puntiforme .Trovare una mutazione puntiforme può anche richiedere sequenziamento del DNA del gene in questione dopo PCR. La tecnica di sequenziamento del DNA prevede l’analisi di ogni base DNA della regione del gene, consentendo il rilevamento di sostituzioni, delezioni o inserzioni, comportano una singola base. Gene sequenziamento è il metodo preferito di prove per Connexin 26 mutazioni (Figura 8; Angeli et al, 2012.).

Figura 8. La diagnosi di connessina 26 mutazioni dal sequenziamento del DNA.
Risultati positivi per la sordità causata da connessina 26. Poiché questo tipo di sordità segue un modello di eredità autosomica recessiva, un paziente affetto dimostra una delezione di una guanina (G) di base in entrambi Connexin 26 (GJB2) geni. Notare le cinque basi (5) G al posto della normale sei (6), scatola rossa.
Oltre alla diagnosi del paziente, tecniche genetiche sono cruciali per l’udienza di ricerca perdite. I geni umani possono essere inseriti in altri organismi di studiare come funzionano. I topi sono gli organismi di scelta nello studio dei geni sordità per le seguenti ragioni: (1) il genoma e orecchio interno del topo sono molto simile a quello umano, (2) topi hanno un breve tempo di gestazione, e (3) sono relativamente facili da accoppiarsi selettivamente. (2012 Angeli et al., Ni et al, 2013). Numerosi modelli di topo che rappresentano modelli in perdita compresa udito umano per la sindrome di Usher e Waardenburg syndrome- sono stati sviluppati.
Sommario
Molti diversi tipi di cambiamenti genetici possono essere associati con la perdita dell’udito. Il tipo di test diagnostico selezionato dipenderà dal tipo di cambiamenti genetici sospetti. Gli indizi possono essere dedotte dal modello storia di famiglia e di successione, ma i medici devono ricordare che una storia familiare negativa non implica che l’eziologia non è genetica. I test genetici può permettere per la diagnosi precoce di altri potenziali problemi di salute, valutare i rischi di ricorrenza, e avvisare gli altri membri della famiglia che possono essere interessati. I test genetici non dovrebbe essere intrapreso fino a cause ambientali, come le infezioni e traumi, sono escluse. Audiologi dovrebbero individuare i genetisti nella loro area e mantenere uno stretto rapporto con loro per la cura ottimale dei pazienti con forme genetiche di perdita dell’udito. Informazioni su sindromi genetiche, le prove, e genetisti nella vostra zona si possono trovare i National Institutes of Health Test genetici Registry e GeneTests siti web.
Ahmed, Z. M., Riazuddin, S., Riazuddin, S., & Wilcox, E. R. (2003). The molecular genetics of Usher syndrome. Clinical Genetics, 63, 431–444.
Angeli, S., Lin, X., & Liu, X. Z. (2012). Genetics of hearing and deafness. Anatomical Record, 295,1812–1829.
Austeng, M. E., Akre, H., Overland, B., Abdelnoor, M., Falkenberg, E. S., & Kvaerner, K. J. (2013). Hearing level in children with Down syndrome at the age of eight. Research in Developmental Disabilities, 34, 2251–2256.
Chen, G., He, F., Fu, S., & Dong, J. (2011). GJB2 and mitochondrial DNA 1555A>G mutations in students with hearing loss in the Hubei province of China. International Journal of Pediatric Otorhinolaryngology, 75, 1156–1159.
de Sousa Andrade, S. M., Monteiro, A. R., Martins, J. H., Alves, M. C., Santos Silva, L. F.,
Quadros, J. M., & Ribeiro, C. A. (2012). Cochlear implant rehabilitation outcomes in Waardenburg syndrome children. International Journal of Pediatric Otorhinolaryngology, 76, 1375–1378.
Evangelidou P., Alexandrou, A., Moutafi, M., Ioannides, M., Antoniou, P., Koumbaris, … Patsalis, P. C. (2013). Implementation of high resolution whole genome array CGH in the prenatal clinical setting: Advantages, challenges, and review of the literature. BioMed Research International, 2013, 346762.
Guha S., Rosenfeld, J. A., Malhotra, A. K., Lee, A. T., Gregersen, P. K., Kane, J. M., … Lencz, T. (2012). Implications for health and disease in the genetic signature of the Ashkenazi Jewish population. Genome Biology, 13, R2.
Hilgert, N., Smith, R. J. H., & Van Camp, G. (2009). Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutation Research, 681, 189–196.
Keats, B. J. (2002). Genes and syndromic hearing loss. Journal of Communication Disorders, 35, 355–366.
Kenneson, A., Van Naarden Braun, K., & Boyle, C. (2002). GJB2 (Connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genetics in Medicine, 4, 258–274.
Kruegel, J., Rubel, D., & Gross, O. (2013). Alport syndrome- insights from basic and clinical research.Nature Reviews Nephrology, 9, 170–178.
Liu, F., Li, P., Liu, Y., Li, W., Wong, F., Du, R., & … Liu, M. (2013). Novel compound heterozygous mutations in MYO7A in a Chinese family with Usher syndrome type 1. Molecular Vision, 19, 695–701.
National Center for Hearing Assessment and Management. (2010). Universal newborn hearing screening: Issues and evidence. Retrieved April 28, 2013.
Ni, C., Zhang, D., Beyer, L. A., Halsey, K. E., Fukui, H., Raphael, Y., & … Hornyak, T. J. (2013). Hearing dysfunction in herterozygous Mitf(Mi-wh)/+ mice, a model for Waardenburg syndrome type 2 and Tietz syndrome. Pigment Cell Melanoma Research, 26, 78–87.
Ouyang, X. M., Hejtmancik, J. F., Jacobson, S. G., Xia, X. J., Li, A., Du, L. L., & … Liu, X. Z. (2003). USH1C: a rare cause of USH1 in a non-Acadian population and a founder effect of the Acadian allele.Clinical Genetics, 63, 150–153.
Ramia, M., Musharrafieh, U., Khaddage, W., & Sabri, A. (2013). Revisiting Down syndrome from the ENT perspective: Review of literature and recommendations [published ahead of print, May 21]. European Archives of Oto-Rhino-Laryngology.
Raut, P., Sriram, B., Yeoh, A., Hee, K. Y., Lim, S. B., & Daniel, M. L. (2011). High prevalence of hearing loss in Down syndrome at first year of life. Annals of the Academy of Medicine, Singapore, 40, 493–498.
Read, A. P., & Newton, V. E. (1997). Waardenburg syndrome. Journal of Medical Genetics, 34, 656–665.
Rehm, H. L. (2005). A genetic approach to the child with sensorineural hearing loss. Seminars in Perinatology, 29, 173–181.
Rheault, M. N. (2012). Women and Alport syndrome. Pediatric Nephrology, 27, 41–46.
Schwartz, S., & Graf, M. D. (2002). Microdeletion syndromes: Characteristics and diagnosis. In Y. S. Fan (Ed.), Molecular cytogenetics: Protocols and applications (pp. 275–290). Retrieved October 3, 2013.
Shott, S. R., Joseph, A., & Heithaus, D. (2001). Hearing loss in children with Down syndrome.International Journal of Pediatric Otorhinolaryngology, 61, 199–205.
Smith, M., & Visootsak, J. (2013). Noninvasive screening tools for Down syndrome: a review.International Journal of Women’s Health, 5, 125–131.
Smith, R. J., Bale, Jr., J. F., & White, K. R. (2005). Sensorineural hearing loss in children. The Lancet, 365, 879–890.
Toriello, H. V., Reardon, W., & Gorlin, R. J. (2004). Hereditary hearing loss and its
syndromes. New York, NY: Oxford University Press.
Van Camp, G., & Smith, R. J. H. (2000). Maternally inherited hearing impairment. Clinical Genetics, 57,409–414.
Van Camp, G., & Smith, R. (2013). Hereditary hearing loss homepage. Retrieved July 31, 2013.
Verma, I. C., Lall, M., & Dua Puri, R. (2012). Down syndrome in India—diagnosis, screening, and prenatal diagnosis. Clinics in Laboratory Medicine, 32, 231–248.
Yan, D., & Liu, X. Z. (2010). Genetics and pathological mechanisms of Usher syndrome. Journal of Human Genetics, 55, 327–335.
Trasmissione autosomica dominante
TRASMISSIONE IPOACUSIA AUTOSOMICA DOMINANTE (AD) (22%)
In un modello di ereditarietà autosomica dominante , un bambino eredita una copia normale di un gene da un genitore e un gene anomalo dall’altro genitore. Nelle famiglie affette da sordità autosomica dominante, uno dei genitori è sordo e porta su un singolo allele del gene la mutazione patogena, che trasmetterà a metà dei suoi figli, che quindi saranno sordi. Il gene anomalo domina il gene normale, così una copia di un gene anormale è sufficiente a causare una malattia autosomica dominante (Fig. 1).La mutazione dell’allele interessato può. per esempio. produrre una proteina anormale, che impedisce la funzione della proteina normale prodotta dall’allele sano. La Sindrome di Waardenburg è la causa più comune di perdita di udito sindromica autosomica dominante, che colpisce 1 su 42.000 persone (Read & Newton, 1997). Quando un genitore ha la sindrome di Waardenburg, ogni bambino di quel genitore ha una probabilità del 50% di essere affetti da sindrome di Waardenburg, assumendo l’altro genitore non ha la sindrome. Il restante 50% della prole in questo accoppiamento sarà inalterato. Caratteristiche tipiche della sindrome di Waardenburg includono perdita dell’udito neurosensoriale, un ciuffo bianco, occhi celesti o di colore diverso, e gli occhi distanziati. Una grande quantità di espressività variabile esiste in Waardenburg sindrome, in modo che i pazienti possono avere una qualsiasi combinazione di caratteristiche, tra cui un udito normale (de Sousa Andrade et al., 2012). Waardenburg sindrome rappresenta circa il 2% dei casi di sordità profonda congenita (de Sousa Andrade et al., 2012).Va notato che, in rari casi, alcuni disturbi autosomica dominante verificarsi a causa di una nuova mutazione nel bambino e nessuno dei genitori ha il disturbo. L’espressività è però variabile in questa modalità di trasmissione: la gravità della ipoacusia può essere variabile tra i vari soggetti colpiti.
Quando l’ipoacusia è sindromica i segni associati alla sindrome possono essere assenti o lievi in alcuni sordi della famiglia e alcuni membri portatori dell’ alterazione genetica possono essere udenti.


Fig.1 ereditarietà autosomica dominante
Le sordità sindromiche rappresentano solo una piccola percentuale di sordità del bambino (10 – 15% circa) e una porzione poco conosciuta, probabilmente inferiore, di sordità dell’adulto. Sono state descritte diverse centinaia di sindromi con sordità (vedi [Gorlin et al.,1995 8]), e a tutt’oggi è stato identificato più di un centinaio di geni. È comunque importante conoscere e ricercare le principali sindromi, poiché la gestione e la valutazione eziologica saranno differenti da una sordità non sindromica. In ragione del grande numero di sindromi rare con sordità qualsiasi patologia malformativa nel bambino deve far effettuare un esame uditivo sistematico. Per le sordità sindromiche inoltre che comprendono una lesione malformativa craniofaciale, la sordità è molto spesso aggravata da un’otite cronica e s’impone un monitoraggio otologico regolare.
Abbiamo elencato nella Tabella 4 le sette sordità sindromiche che ci sembrano più importanti e che devono essere conosciute dai diversi specialisti che assumono in carico le sordità, a motivo della loro frequenza e/o della loro gravità potenziale. La Tabella 5 elenca in modo più completo le sindromi no eccezionali i cui geni sono stati identificati (per una rassegna esauriente dei geni clonati della sordità sindromica, vedi [Marlin S. 2004 9]). Descriveremo qui le sette sindromi della Tabella 1.
Tre sindromi autosomiche dominanti
Devono essere conosciute dagli otorinolaringoiatri (ORL) poiché sono frequenti e probabilmente sottodiagnosticate: la sindrome di Waardernburg, la sindrome branchio-oto-renale (BOR) e la sindrome di Stickler.
Associa una sordità ad anomalie della pigmentazione dovute a un’assenza di melanociti in diversi organi. Ciò può riguardare i capelli (ciocche bianche) e le sopracciglia, gli occhi (occhi molto azzurri, depigmentati, depigmentazione sul fondo dell’occhio), la cute (macchie cutanee). In alcune forme (Waardernburg di tipo 1 e 3), è presente un’anomala distanza tra gli occhi, con i canti interni spostati verso l’esterno con riduzione di lunghezza della fessura palpebrale (distopia cantale). La sordità è molto variabile, mono- o bilaterale, da leggera a profonda (profonda nel 35% dei casi), [Hildesheimer et al.,1989 24] con o senza malformazione dell’orecchio interno. La trappola per la diagnosi è che le particolarità fisiche che possono essere presenti nella famiglia dei sordi ed essere molto suggestive non sono spontaneamente descritte all’anamnesi, non essendo considerate patologie. Le ciocche bianche sono inoltre il più delle volte tinte. La domanda circa la presenza di occhi depigmentati o di ciocche bianche nella famiglia deve quindi essere posta sistematicamente. Sono stati descritti quattro tipi clinici di sindrome di Waardernburg in funzione dei segni associati:
• il tipo I è associato a una distopia cantale;
• il tipo II, il più frequente, è privo di distopia cantale;
• il tipo III (o Klein-Waardenburg) è un tipo I associato a malformazioni delle estremità;
• il tipo IV è un tipo II con malattia di Hirschsprung. Attualmente, sono stati identificati sei geni, onnicomprensivi nella Tabella 4. Essi non rendono conto di tutte le sindromi di Waardernburg (per esempio, PAX3 è chiamato in causa in tre quarti dei tipi I e MITF nel 15% dei tipi II soltanto). [Tassabehjiet al.,1995 25]
Sindrome branchio-oto-renale
La sindrome branchio-oto-renale (BOR) associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000. [Fraser et al.,1980 26] Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un’interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un’ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell’orecchio esterno (malformazioni del padiglione, aplasia dell’orecchio, encondromi, stenosi dei condotti uditivi), dell’orecchio medio (esiste una componente di trasmissione all’audiogramma) e dell’orecchio interno (varie malformazioni cocleovestibolari). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi). In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell’orecchio esterno, è consigliabile eseguire un’ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1. [Abdelhak et al.,1997;, Ruf et al.,2004 28] Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale. [Vincent et al., 1997 29]
Sindrome di Stickler
Questa sindrome è dovuta a una alterazione delle catene ![]() di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
La sordità di percezione, trasmissione o mista è incostante, e spesso mascherata o aggravata dai problemi di otite cronica che sono associati all’incompetenza faringea. Essa è spesso evolutiva. Sono descritti tre tipi di sindrome di Stickler: nei tipi 1 e 3, agli altri elementi della sindrome è associata una miopia molto forte con rischio di degenerazione vitro-retinica. L’esame oftalmologico di ogni bambino sordo e di ogni lattante con incompetenza faringolaringea deve permettere d’individuare questa sindrome e correggere precocemente la miopia.
Gorlin R.J., Toriello H.V., Cohen M.M. Hereditary hearing loss and its syndromes New York: Oxford University Press (1995).
Marlin S. Surdités génétiques Génétique médicale Paris: Masson (2004). 281-299
Hildesheimer M., Maayan Z., Muchnik C., Rubinstein M., Goodman R.M. Auditory and vestibular findings in Waardenburg’s type II syndrome J. Laryngol. Otol. 1989 ; 103 : 1130-1133
Tassabehji M., Newton V.E., Liu X.Z., Brady A., Donnai D., Krajewska-Walasek M., e al. The mutational spectrum in Waardenburg syndrome Hum. Mol. Genet. 1995 ; 4 : 2131-2137
Fraser F.C., Sproule J.R., Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss Am. J. Med. Genet. 1980 ; 7 : 341-349 [cross-ref]
Abdelhak S., Kalatzis V., Heilig R., Compain S., Samson D., Vincent C., e al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family Nat. Genet. 1997 ; 15 : 157-164 [cross-ref]
Ruf R.G., Xu P.X., Silvius D., Otto E.A., Beekmann F., Muerb U.T., e al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes Proc. Natl. Acad. Sci. USA 2004 ; 101 : 8090-8095 [cross-ref]
Vincent C., Kalatzis V., Abdelhak S., Chaib H., Compain S., Helias J., e al. BOR and BO syndromes are allelic defects of EYA1 Eur. J. Hum. Genet. 1997 ; 5 : 242-246
APPROFONDIMENTO
Waardenburg sindrome (anche sindrome di Shah Waardenburg, sindrome di Waardenburg-Klein, sindrome di Mende II, Van der sindrome Hoeve-Halbertsma-Waardenburg, sindrome di ptosi-epicanto, Van der sindrome Hoeve-Halbertsma-Gualdi, tipo Waardenburg Pierpont, [5] Van der Hoeve sindrome Waardenburg-Klein, sindrome di Waardenburg II e sindrome di Vogt) è una rara malattia genetica più spesso caratterizzato da diversi gradi di sordità , difetti minori nelle strutture derivanti dalla cresta neurale , e anomalie della pigmentazione. Disagi Nella miogenesi , in particolare mutazioni in Pax3, possono causare la sindrome di Waardenburg I e III.E ‘stata descritta per la prima nel 1951.

 Che cosa è la sindrome di Waardenburg?,
Che cosa è la sindrome di Waardenburg?,
Le cause della sindrome di Waardenburg,
Fisiopatologia,
Epidemiologia,
Diagnosi e valutazione audiologica,
Che cosa è la sindrome di Waardenburg?
Waardenburg sindrome è una malattia rara caratterizzata da sordità in associazione con anomalie della pigmentazione e difetti dei tessuti cresta di derivazione neurale.
Sindrome è una parola che descrive un gruppo di caratteristiche fisiche che si verificano insieme. Le persone che hanno WS di solito hanno un aspetto unico. Questa unicità è causato da cambiamenti nel loro corredo genetico.
Sindrome di Waardenburg è una condizione genetica che causa una persona di essere nato con le caratteristiche facciali inusuali e perdita dell’udito. Questi problemi fisici possono comparire nel corso della vita della persona. Non tutti con WS avrà tutte le caratteristiche della sindrome, e non tutti avranno una perdita uditiva, ma vi è una maggiore probabilità che lo faranno.
Hammerschlag, nel 1907, e Urbantschitsch, nel 1910, entrambi citano eterocromia dell’iride, albinismo parziale con complicanze di sordomutismo Nel 1916, van der Hoeve descrive una distopia angoli palpebrali mediale lateroversa in una coppia di gemelle monozigoti con Sordomutismo La Sindrome di Waardenburg è stato identificato nel 1951 da Petrus Johannes Waardenburg(1886-1979), un oculista olandese che ha notato che molti dei suoi pazienti avevano caratteristiche facciali uniche, occhi di colore diverso avevano pure, spesso, la perdita dell’udito. In effetti, una caratteristica primaria di Waardenburg è perdita di udito – insieme con gli occhi blu/ marroni o due brillanti occhi azzurri. Un’altra caratteristica facilmente riconoscibile è capelli bianchi sulla testa. Da allora, sono stati descritti quattro tipi specifici di WS. La condizione ha descritto è ora classificato come WS1.
ha definito la sindrome con le seguenti 6 principali caratteristiche
· Spostamento laterale della angoli palpebrali mediali combinata con distopia del punto lacrimale e blefarofimosi
· Aspetto di Eyes Wide-set a causa di una prominente, larga radice del naso(distopia dei canti ),associata in particolare con il tipo I),nota anche come telecanto ;
· Ipertricosi della parte mediale delle sopracciglia che toccano nel mezzo
· Un ciuffo di capelli bianchi ( poliosi ), o ingrigimento precoce dei capelli;
· Occhi molto chiari o azzurri brillanti, occhi di due colori diversi (eterocromia completa ), o gli occhi con un’iride con due colori diversi (eterocromia settoriale);
· Sordità da moderata a profonda (frequenza più alta associata al tipo II);
L’oculista svizzero David Klein, nel 1947, ha anche fatto contributi per la comprensione della sindrome,Klein ha riportato il caso di una ragazza di 10 anni con deafmutism, albinismo parziale della pelle e dei capelli, iridis ipocromia, blefarofimosi con ipertelorismo e l’assenza dell’angolo nasofrontal, ipertricosi delle sopracciglia, e molteplici anomalie associate (mio -osteo-Articulare displasia).
WS2 è stato identificato nel 1971, per descrivere i casi di ” distopia dei canti “non era presente. [4] WS2 è ora diviso in sottotipi, basato sul gene responsabile.
Sono stati identificati altri tipi, ma sono meno comuni.
Mutazioni nel EDN3, EDNRB, MITF, PAX3, SNAI2, e SOX10 sono i geni che sono affetti dalla sindrome di Waardenburg. Alcuni di questi geni sono coinvolti nella realizzazione dei melanociti, che rende il pigmento melanina. La melanina è un pigmento importante nello sviluppo di capelli, il colore degli occhi, della pelle, e le funzioni dell’orecchio interno. Così la mutazione di questi geni può portare a pigmentazione anomala e la perdita dell’udito. [5] PAX3 e MTIF mutazione del gene avviene in tipo I e II (WS1 e WS2). Tipo III (WS3) mostra mutazioni del gene PAX3 anche. Mutazioni del gene SOX10, EDN3, o EDNRB si verificano in tipo IV.Tipo IV (WS4) può anche interessare porzioni di sviluppo delle cellule nervose che potenzialmente possono portare a problemi intestinali.
Le cause della sindrome di Waardenburg
Questa condizione è solitamente ereditata come autosomica dominante modello, il che significa una copia del gene alterato è sufficiente a causare il disturbo. Nella maggior parte dei casi, una persona affetta ha un genitore con la condizione. Una piccola percentuale di casi derivano da nuove mutazioni nel gene; questi casi si verificano in persone senza storia di malattia nella loro famiglia.
Alcuni casi di tipo II e sindrome tipo IV Waardenburg sembrano avere un autosomica recessiva modello di ereditarietà, il che significa due copie del gene devono essere modificati per una persona di essere colpiti dalla malattia. Molto spesso, i genitori di un bambino con una malattia autosomica recessiva non sono interessati, ma sono portatori di una sola copia del gene alterato.
· 
Waardenburg sindrome è solitamente ereditata come modello autosomico dominante .
· 
Tipi II e sindrome IV Waardenburg possono talvolta avere un modello di ereditarietà autosomico recessivo .
I geni sono responsabili per dirigere la crescita e lo sviluppo degli esseri umani. Quando i geni sono mutati, o diverso dal normale, possono causare cambiamenti nella normale crescita e sviluppo. In WS, i geni mutati sono responsabili di distribuzione del pigmento nel corpo di sviluppo embrionale e fetale. Questi geni possono causare pigmento da distribuire in modo anomalo. Quando questo accade, alcune zone del corpo possono avere troppo o troppo poco pigmento, o nessun pigmento affatto. Alterazioni della pigmentazione possono produrre occhio inusuale, la pelle, o il colore dei capelli così come la sordità. Gli scienziati non hanno ancora capito esattamente che cosa causa le mutazioni genetiche che producono WS, anche se sono stati identificati i geni stessi.
Fisiopatologia
La sindrome Waardenburg è una malattia rara con una modalità autosomica dominante. Diverse ipotesi sono state avanzate per spiegare tutte le caratteristiche cliniche della sindrome.
-
- La teoria della cresta neurale carente, suggerisce una anomalia dello sviluppo della cresta neurale come causa di malattia: L’associazione della sindrome di Waardenburg e megacolon congenito aganglionico supporta questa ipotesi.
-
- La sindrome di Waardenburg come una parte della sindrome del primo arco
-
- Un rapporto della sindrome di Waardenburg con lo status dysraphicus
- La teoria della necrosi intrauterina
Nessuna di queste possibilità spiega tutte le caratteristiche della sindrome di Waardenburg. Cause ereditarie rappresentano circa il 50% degli individui osservati per l’infanzia (prelinguale) perdita di udito, di cui il 70% sono dovuti a mutazioni in numerosi geni singoli che compromettono la funzione uditiva solo (non sindromica). Il resto sono associate ad altre anomalie dello sviluppo chiamati sordità sindromica.
I Geni responsabili di forme sindromiche di perdita dell’udito nella sindrome di Waardenburg includono PAX3 sulla banda 2q37, osservata nei tipi I e III, e MITFmappato su 3p12-p 14,1 per il tipo II(Tachibana M.,1997; Zhang H et al., 2012; Wildhardt G et al 2013 . La sindrome di Waardenburg è autosomica dominante per la maggior parte delle persone con tipo I, II, e III. La sindrome di Waardenburg di tipo IV è autosomica recessiva con penetranza variabile ed è causa di mutazioni del gene SOX10 o del recettore dell’endotelina-B ( EDNRB ), che sembrano correlare con l’intestinale e / o sintomi neurologici manifesta nei pazienti(Morell R, et al.1998;DeStefano AL, et al.,1998;Bondurand N, et al.,2000; Dundar M, et al.,2001;Chan KK, et al.,2003; Sznajer et al.,2008; Oshimo T, et al.,2012;Jung HJ, et al.,2013;hanno descritto un sito di splice mutazione SOX10 (c.698-2A> C), che ha provocato una sindrome di Waardenburg grave di tipo 4 senza malattia di Hirschsprung . Il bambino presentava vivaci occhi blu, ritardo mentale, sinofria, sordità, canali completi bilaterali semicircolari, e neuropatia periferica.
Epidemiologia
Quanto rara è la sindrome di Waardenburg?
La frequenza della sindrome Waardenburg è stimato a 1 caso ogni 212.000 persone nella popolazione generale dei Paesi Bassi, ma a causa di una bassa penetranza di circa il 20%, la frequenza dell’intero sindrome (con o senza sordità) è probabilmente di circa 1 caso per ogni 42.000 persone. In Kenya, Secondo il National Institutes of Health, un individuo su 10.000 a 20.000 avrà la sindrome di Waardenburg. Inoltre, dal momento che i primi due tipi (tipo I e II) sono più comuni e sono associati con la perdita dell’udito, si può presumere che la maggior parte degli individui con Waardenburg avrà la perdita dell’udito.
La sindrome è stata osservata in 0,9-2,8% delle persone con sordomutismo.
Mortalità / morbilità
I bambini con sindrome di Waardenburg hanno una aspettativa di vita normale. La morbilità è legato alla sordità e difetti dei tessuti cresta di derivazione neurale, tra cui ritardo mentale, convulsioni, disturbi psichiatrici, anomalie scheletriche, e disturbi oculari (cataratta comprese).
Razze
Waardenburg sindrome colpisce persone di tutte le razze di tutto il mondo.
Sesso
La malattia colpisce ugualmente entrambi i sessi. Non sono stati trovati differenze sessuali tra persone con sordomutismo congenito.
Età
Come una malattia ereditaria, la sindrome di Waardenburg può essere riconosciuto immediatamente o subito dopo la nascita. Alcune caratteristiche dermatologiche (ad esempio, poliosi) cambiano con l’età.
Chi è a rischio?
Si stima che circa 1 su 4000 individui nascono con WS. WS si verifica in tutte le razze, e si verifica in modo uguale in maschi e femmine. Nella maggior parte dei tipi di WS, c’è una probabilità del 50% che un bambino nato da una madre con un gene normale e un genitore con il gene WS avrà WS. Chiunque può nascere con WS come risultato di una nuova mutazione genetica.
Il trattamento dipende dal fenotipo dell’individuo con Sindrome di Waardenburg. È possibile ottenere un trattamento per uno dei seguenti segni fenotipiche:
1) Sordità da moderata a profonda (frequenza più alta associata al tipo II);
2) La malattia di Hirschsprung (HD)
3) problemi estetici
4) palatoschisi
5) Mano e contratture delle dita
Il trattamento per queste caratteristiche comporta la consulenza genetica e / o rinvio di appropriarsi medici professionisti come un audiologo, logopedista, fisioterapista e chirurgo plastico.
Diagnosi e valutazione audiologica
Americans with Disabilities Act (ADA)
La perdita dell’udito può essere trattata, l’udito deve essere controllato da un audiologo. Lui o lei determinerà il grado e il tipo di perdita uditiva. Ecco un elenco di alcuni dei test che un audiologo può somministrare. Non tutti hanno bisogno di tutti questi test. Il medico o l’audiologo può raccomandare quelli necessari, in base alla storia familiare ei sintomi della perdita dell’udito.
Valutazione audiologica
1. Otoscopia
 L’Otoscopia è un esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di infezione.
L’Otoscopia è un esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di infezione.
2. Timpanometria
 La timpanometria è una misura della rigidità del timpano e ci dice come gli ossicini dell’orecchio medio funzionano. Questo test consente di rilevare liquido nell’orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si trovano nell’orecchio medio, un perforazione nel timpano, e una malattia dell’orecchio medio chiamato otosclerosi. L’audiologo metterà una sonda morbida nel canale uditivo e una macchina rilascia una piccola quantità di pressione. Il movimento strumento misura le risposte del timpano alle variazioni di pressione e registra il risultato su un grafico.
La timpanometria è una misura della rigidità del timpano e ci dice come gli ossicini dell’orecchio medio funzionano. Questo test consente di rilevare liquido nell’orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si trovano nell’orecchio medio, un perforazione nel timpano, e una malattia dell’orecchio medio chiamato otosclerosi. L’audiologo metterà una sonda morbida nel canale uditivo e una macchina rilascia una piccola quantità di pressione. Il movimento strumento misura le risposte del timpano alle variazioni di pressione e registra il risultato su un grafico.
3. Audiometria Tonale ai Toni Puri
 Il test ai toni puri è il tipo più comune di valutazione dell’udito. L’audiologo può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il suono. L’audiologo registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri comprende anche il test di conduzione per via ossea. Durante questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e determinare quale parte dell’orecchio è responsabile di qualsiasi perdita uditiva.
Il test ai toni puri è il tipo più comune di valutazione dell’udito. L’audiologo può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il suono. L’audiologo registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri comprende anche il test di conduzione per via ossea. Durante questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e determinare quale parte dell’orecchio è responsabile di qualsiasi perdita uditiva.
4. Emissioni Otoacoustiche (OAE)
La coclea è la parte dell’orecchio che riceve informazioni dai suoni in entrata e invia i suoni al cervello. La coclea contiene molte terminazioni nervose chiamate “cellule ciliate”. Quando questi terminazioni nervose lavorano correttamente, i suoni vengono inviati al cervello in maniera accurata ed il cervello interpreta questi suoni come la musica, la parola, o il rumore. Le coclee normali hanno le cellule ciliate esterne che producono rumore. Il test’s OAE registra i suoni che l’orecchio produce e aiuta l’audiologo a determinare se il vostro orecchio interno trasmette il suono al cervello.
5. Auditory Brainstem Response (ABR)
Il test ABR è uno strumento diagnostico non invasivo che misura le onde cerebrali in risposta ad uno stimolo uditivo. Esso misura l’attività della via uditiva che si trova nel vostro tronco encefaloo Per ulteriori informazioni sui test ABR, andare a questi siti.:
Le prove di cui sopra sono solo alcuni dei possibili modi in cui gli audiologi determinano se l’udito è normale. Per saperne di più sulla diagnosi della perdita dell’udito, visitare i seguenti siti web:
American Speech and Hearing Association
Le opzioni per il trattamento WS variano a seconda delle particolari caratteristiche WS. Parte di udito trattamento di perdita è l’intervento di un consulente genetico. Il trattamento può includere uno o una combinazione delle seguenti opzioni. Ricordate che il trattamento (s) scelto da una persona con WS, non può essere adatto per voi o il vostro bambino.
1. Genetic Testing & Counseling
2. Opzioni per il trattamento di perdita di udito includono:
-
- Apparecchi acustici o Amplification
-
- Impianti cocleari
-
- Sign Language
- Logoterapia
Voi o il vostro bambino può scegliere tra una varietà di scuole per sentire gli individui con problemi. Considerate le esigenze specifiche del vostro bambino e il grado di perdita di udito quando si sceglie una scuola. Molte scuole specializzate in formazione per le persone con problemi di udito. Alcune scuole si affidano completamente sulla lingua dei segni americana, alcuni includono una combinazione del linguaggio parlato e segni orale, e altri usano il discorso solo orale. Oltre alle scuole di specializzazione, si può scegliere di inviare il vostro bambino a una scuola pubblica all’interno della vostra comunità. ‘Gli Stati Uniti individui con disabilità Education Act (IDEA) afferma che “ogni bambino ha diritto a una istruzione gratuita e adeguata nel contesto meno restrittivo.” In altre parole, il bambino deve essere dotato delle risorse adeguate in una scuola vicino te.
Caratteristiche fisiche
Caratteristiche fisiche dei WS sono classificati come maggiore o minore. Per essere diagnosticati con WS una persona deve dimostrare due caratteristiche principali, o una maggiore e due minori caratteristiche della sindrome. Non tutti coloro che hanno WS avrà tutte queste caratteristiche, e non tutte le caratteristiche della sindrome sarà presente alla nascita. Alcune caratteristiche si sviluppano più tardi nella vita, e alcuni diventano più pronunciati con l’età.
Le principali caratteristiche sono elencate di seguito. Le descrizioni di ciascuna si trovano qui .
• Eterocromia Iridis
• luminosi occhi azzurri
• Distopia dei canti (spostamento laterale angoli interni palpebrali )
• Broad, radice del naso prominente
• Piccolo mid-face
• capelli prematuramente grigi
• sordità neurosensoriale congenita
• WS Tipo 4 & Malattia di Hirschsprung
Eterocromia Iridis
|
|
|

Occhi molto chiari o brillanti azzurri, occhi di due colori diversi (eterocromia completa), o gli occhi con un’ iride con due colori diversi (eterocromia settoriale);”Eterocromia Iridis” significa che qualcuno, o un animale, ha gli occhi di due colori diversi, come fa il gatto nella foto a destra. Può anche significare due colori nella stessa dell’occhio, come nella foto a sinistra. Questa persona è nata con gli occhi azzurri, e sviluppato macchie di marrone durante l’adolescenza.
Eterocromia rende occhi sguardo insolito, ma non influenza la visione della persona. WS solito non causare problemi di visione.
È interessante notare che gli animali, tra cui cavalli, cani, topi e gatti possono avere WS. Animali sordi fanno i buoni animali domestici, ma sono spesso distrutti nella convinzione che essi non possono essere addestrati. Animali sordi possono fare buoni animali domestici, se sono tenuti in un ambiente protetto.
|
|
|


Le persone con WS hanno spesso occhi blu brillanti belli, in combinazione con una o più delle altre caratteristiche di WS. Occhi azzurri e prematuramente capelli grigi si verificano spesso in combinazione, come fanno nella foto a sinistra. Qualsiasi persona con gli occhi azzurri può avvertire fotofobia, difficoltà di vedere alla luce del sole, perché le persone con gli occhi azzurri non hanno lo strato sovrastante di pigmento scuro che le persone con marrone, nocciola, o gli occhi verdi fanno. Questo pigmento aggiuntivo protegge gli occhi dalla luce forte. Le persone con gli occhi azzurri anche spesso hanno scarsa visione notturna. Gli occhi azzurri da soli non sono diagnostici di WS; tuttavia, gli occhi azzurri in combinazione con i capelli precocemente grigi, aree di pelle depigmentazione, perdita dell’udito suggeriscono WS.
|
|
|


La “Distopia dei canti” descrive l’aspetto degli occhi della persona e ponte nasale. Le persone con distopia dei canti hanno ampie, ponti nasali piatte, con pieghe di pelle che copre angoli interni degli occhi. Queste pieghe della pelle danno un aspetto asiatico agli occhi di persone con WS. Persone asiatici come quello nella foto a destra, hanno pieghe cutanee copre gli angoli interni degli occhi chiamato epicanto. Queste pieghe sono una caratteristica del viso normale causata dai ponti nasali pianeggianti tipici di persone asiatiche. Dystopia dei canti, come mostrato nella foto a fianco, non è una condizione normale per persone di ogni razza. I medici utilizzano una formula denominata “W Index” per calcolare la distanza tra gli occhi per stabilire se qualcuno ha distopia dei canti.
Broad nasale Root
 Broad, importanti radici nasali si trovano spesso nelle persone con WS. La radice nasale è l’area tra gli occhi del ponte della occhiali poggia. Le persone con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e caratteristici nevi di WS.
Broad, importanti radici nasali si trovano spesso nelle persone con WS. La radice nasale è l’area tra gli occhi del ponte della occhiali poggia. Le persone con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e caratteristici nevi di WS.
Piccola Mid-Face
 Le persone con WS possono avere ossa facciali che sono più piccoli del normale. Queste ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le occhiaie. Quando ossa metà viso sono più piccolo del previsto, il viso della persona può apparire piatta nel profilo. Ossa nasali appiattite possono causare gli occhi della persona a comparire ampiamente impostare e “orientale-looking”, anche se la persona non è asiatico.
Le persone con WS possono avere ossa facciali che sono più piccoli del normale. Queste ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le occhiaie. Quando ossa metà viso sono più piccolo del previsto, il viso della persona può apparire piatta nel profilo. Ossa nasali appiattite possono causare gli occhi della persona a comparire ampiamente impostare e “orientale-looking”, anche se la persona non è asiatico.
Quest’uomo ha piccole ossa della faccia media, eterocromia dell’iridis e capelli precocemente grigi – tutte le caratteristiche di WS.
Capelli prematuramente grigi, o macchie di capelli grigi nei capelli di colore più scuro sono anche segni di WS. Molte persone con WS hanno una striscia bianca di capelli al centro della loro fronte. Altri possono avere i capelli che si è interamente grigio quando erano adolescenti. Alcune persone con WS hanno ciglia e sopracciglia bianche. I capelli grigi si verifica spesso in combinazione con gli occhi blu e la perdita dell’udito. Persone i cui capelli sono diventati grigi prematuramente possono scegliere di tingersi i capelli, quindi questa funzione non è sempre visibile in qualcuno con WS. L’uomo nella foto (in alto a destra) ha i capelli che sono diventati grigi quando era adolescente.
Sordità neurosensoriale
 Ipoacusia neurosensoriale può essere presente alla nascita (congenita), o può apparire più tardi nella vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita dell’udito in un solo orecchio, e alcuni individui hanno perdite uditive molto lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell’udito non è una caratteristica visibile, le persone che hanno alcune delle altre caratteristiche di WS devono sempre ricevere una valutazione dell’udito. Un ragazzo con un impianto cocleare è raffigurato qui sotto.
Ipoacusia neurosensoriale può essere presente alla nascita (congenita), o può apparire più tardi nella vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita dell’udito in un solo orecchio, e alcuni individui hanno perdite uditive molto lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell’udito non è una caratteristica visibile, le persone che hanno alcune delle altre caratteristiche di WS devono sempre ricevere una valutazione dell’udito. Un ragazzo con un impianto cocleare è raffigurato qui sotto.
Caratteristiche minori di WS
Caratteristiche di minore importanza sono i seguenti, con le descrizioni disponibili qui :
• leucoderma congenita
• Diverse nevi (moli)
• anomalie sopracciglio
• ipoplasico nasale alae
• Sindattilia
• mento sfuggente
• labio-palatoschisi
• Spina bifida
• Disturbi della comunicazione
• Camptodattilia
• caratteristiche rare

Le persone con WS volte hanno “leucoderma congenita”, o l’assenza di pigmento nella loro pelle. Perché queste macchie bianche o chiare di pelle non hanno il pigmento, si scottature facilmente e sono a rischio di sviluppare il cancro della pelle. Leucoderma è facilmente visibile sullepersone con la pelle scura, e può causare problemi estetici o sociali per queste persone. Ecco una foto di leucoderma su una mano.

Nei Multipli
I nei sono raccolte di cellule pigmentate. I nei sono spesso dispersi attraverso la pelle di persone con WS, solitamente nelle zone della testa e del collo, come nella donna in questa fotografia. I nei dovreb-
bero essere controllati, per essere certi che non diventino maligni, e se necessario dovrebbe essere rimosso chirurgicamente.

Eyebrow Anomalie
Anomalie del sopracciglio sono comuni nelle persone con WS. Le sopracciglia possono essere depigmentate e possono crescere attraverso il ponte del naso e riunite sulla linea mediana. Questa condizione si chiama “synophrys,” o unibrow. Sopracciglia folte o svasate possono creare un viso con aspetto insolito. Nella foto a sinistra,vengono visualizzati sul ragazzo Sopracciglia svasatE
 “Ipoplasia” indica sottosviluppati. Le persone con WS possono avere ipoplasia dei muscoli della spalla e del collo, così come sottosviluppo delle ossa della faccia media. Sono comuni anche ali nasali ipoplasiche (ai lati della punta del naso) nelle persone con WS.
“Ipoplasia” indica sottosviluppati. Le persone con WS possono avere ipoplasia dei muscoli della spalla e del collo, così come sottosviluppo delle ossa della faccia media. Sono comuni anche ali nasali ipoplasiche (ai lati della punta del naso) nelle persone con WS.
Questa donna ha ali nasali ipoplasiche ed ipoplasia dei muscoli della spalla, così come suo figlio. Ha anche eterocromia iridis e nevi multipla, visibile in questa fotografia.
Sindattilia
 Le persone con le dita dei piedi fusi hanno una condizione chiamata “sindattilia.” Sindattilia e polidattilia (troppe dita delle mani o dei piedi) si trovano talvolta nelle persone con WS. Dita fuse sono raffigurati a sinistra.
Le persone con le dita dei piedi fusi hanno una condizione chiamata “sindattilia.” Sindattilia e polidattilia (troppe dita delle mani o dei piedi) si trovano talvolta nelle persone con WS. Dita fuse sono raffigurati a sinistra.

Stempiato Chin
“Mandibola retrognatica” è un altro nome per una piccola mascella inferiore, o mento sfuggente. Le persone con WS possono avere retrognazia nonché orecchie inclinate con inclinazione verso la parte posteriore della testa. La mascella di questa donna è retrognatica (foto a destra). È anche possibile vedere nevi vicino al naso e sul collo.
Labbro leporino e palatoschisi
 La palatoschisi “accade quando il tetto della bocca non forma correttamente in utero. Un palato che non si è fuso è chiamato palatoschisi. Un labbro che non si è fuso è chiamato labbro leporino. Labio e / o palatoschisi sono talvolta presente nei neonati che hanno anche WS. Queste condizioni devono essere corrette chirurgicamente in modo che la persona possa imparare a mangiare e parlare normalmente.
La palatoschisi “accade quando il tetto della bocca non forma correttamente in utero. Un palato che non si è fuso è chiamato palatoschisi. Un labbro che non si è fuso è chiamato labbro leporino. Labio e / o palatoschisi sono talvolta presente nei neonati che hanno anche WS. Queste condizioni devono essere corrette chirurgicamente in modo che la persona possa imparare a mangiare e parlare normalmente.
Questa donna è nata con il labbro leporino e palatoschisi e ora ha un volto normale ricerca e ottime capacità di linguaggio.
Per ulteriori informazioni, consultare il americana palatoschisi – cranio Association (ACPCA) sito web.
“Spina bifida,” o della colonna vertebrale aperto, a volte si verifica nei bambini che hanno WS. Spina bifida si verifica quando le ossa della colonna vertebrale non si fondono durante lo sviluppo fetale. Il fluido cerebrospinale (CSF)che è contenuto del midollo spinale può essere spinti attraverso l’apertura verso l’esterno del corpo. Ciò può verificarsi in qualsiasi punto lungo la lunghezza della colonna vertebrale, dalla base del collo, alla base della colonna vertebrale. La spina bifida deve essere corretto subito dopo la nascita per prevenire l’infezione del midollo spinale e del sistema nervoso.

Persone che hanno WS e perdita di udito, palatoschisi o labbro leporino possono avere difficoltà ad imparare a parlare o usare un linguaggio appropriato. Intervento precoce offre la migliore possibilità per questi bambini di imparare a comunicare in modo efficace. Persone che hanno WS di solito non sono cognitivamente compromesse o ritardati mentali. I loro problemi del linguaggio e, se del caso, il risultato di essere in grado di sentire il discorso normale e il linguaggio, e raramente, dalle labbra e palati schisi non riparati. Alcune persone che hanno WS e sorde, preferiscono far parte della comunità dei sordi e imparare a comunicare attraverso il linguaggio dei segni. Altre persone non udenti scelgono di usare apparecchi acustici o impianti cocleari per consentire loro di imparare la lingua e la parola orale.
“Camptodattilia” significa diti che sono curvati verso l’alto e permanentemente flesse, come le dita di questo individuo che ha WS (a destra). La camptodattilia a volte è trattata con la terapia fisica e chirurgia correttiva.
Molto raramente, gli individui con WS possono nascere senza occhi, una condizione chiamata “anoftalmia.” Questo è più probabile che accada se i genitori del bambino erano stretti consanguinei (accoppiamento tra consanguinei). Alcuni bambini nati da accoppiamenti tra consanguinei sono nati con WS e difetti degli arti. Questo è molto rara combinazione di caratteristiche fisiche. Il ritardo mentale è stata osservata in questi bambini, ma è incerto se i problemi cognitivi derivano da WS o da qualche altra causa.
Molte condizioni hanno sintomi simili, e WS non fa eccezione. L’unico modo assolutamente preciso per sapere se qualcuno ha WS è di test genetici. Tuttavia, i test genetici non è ancora disponibile per tutti i tipi di WS. Se pensate che voi o qualcuno che conosci ha WS, assicuratevi di cercare un modello di anomalie, non solo una o due cose.
Alcune altre condizioni hanno caratteristiche che si trovano anche in WS. Queste condizioni di solito causano problemi di pigmento nei capelli, pelle e / o degli occhi. “L’albinismo” si intende la mancanza di pigmento nella pelle, capelli o gli occhi. Un certo numero di problemi fisici causano l’albinismo, alcuni sono ereditarie (genetiche), e alcuni non lo sono. Le descrizioni delle condizioni elencate di seguito sono qui .
· Piebaldism
· Oculocutaneous Albinismo / Oculare
· Vitiligine
· Sindrome di Horner
· Trauma Eye
“Piebaldismo” è una condizione autosomica dominante che è stata fatta risalire a una mutazione del gene specifico. Le persone che hanno piebaldismo sono molto simili a chi ha l’WS. Essi possono avere zone di pelle depigmentazione sulla loro testa e del tronco, nonché sulla loro sopracciglia, palpebre, ciglia e capelli. Essi possono anche avere occhi di due colori diversi (Eterocromia Iridis). La sordità non è caratteristica di piebaldismo, né ampia radice del naso o distopia dei canti. Piebaldismo si trova spesso nei pazienti di origine africana. E ‘impossibile capire la differenza tra piebaldismo e WS solo guardando l’aspetto fisico di qualcuno. Conoscere la storia della perdita di udito famiglia può essere utile nel fare la distinzione, ma l’analisi genetica è l’unico modo di distinguere tra le due condizioni.
Albinismo Oculocutaneo / Oculare
Le persone con “albinismo oculocutaneo” possono avere totale assenza di pigmento nella loro pelle, capelli e occhi. A volte il pigmento manca solo dai loro occhi. In questo caso la persona ha una condizione chiamata “albinismo oculare.” Il paziente può avere fotofobia, strabismo e riduzione del visus. Alcuni pazienti hanno anche nistagmo congenito (oscillazione involontaria dei bulbi oculari). La maggior parte delle persone affette da albinismo oculare hanno una condizione genetica legata al cromosoma X denominata Nettleship-Falls albinismo oculare. Ciò significa che il gene che causa albinismo oculare che si trova sul cromosoma X e viene trasmesso dalla madre al figlio. La forma autosomica recessiva meno comune dell’albinismo oculare è una specifica mutazione genetica e colpisce maschi e femmine in ugual misura.
Le persone con albinismo oculocutaneo tipo 2 sono simili a persone con albinismo oculocutaneo, salvo che la loro pelle e capelli hanno una diminuzione generalizzata di pigmento. Le persone con questo problema hanno la pelle pallida e capelli colorati giallastro. L’Albinismo oculocutaneo può essere presente in WS2, ma sono necessario i test genetici per una diagnosi differenziale accurata, perché l’albinismo oculocutaneo avviene in molte altre sindromi.
L’Organizzazione Nazionale di albinismo e Ipopigmentazione (NOAH) fornisce informazioni e sostegno alle persone che hanno condizioni di pigmentazione. Possono essere contattati al http://www.albinism.org , o PO Box 959 East Hampstead, NH 03.826-0.959.
La “Vitiligine” è una malattia autoimmune causata da mutazioni genetiche sui cromosomi 1, 7, 8 e 4 persone con vitiligine hanno chiazze di depigmentazione sulla pelle, sul viso, mani, piedi, gomiti, ginocchia e torace. I loro capelli possono perdere il pigmento, e le aree depigmentate della pelle possono aumentare di dimensioni. La perdita dell’udito e anomalie cranio-facciali non sono associati con vitiligine. Alcuni casi di vitiligine sembrano essere ereditarie, mentre altri sono associati a trauma, ipertiroidismo, diabete e problemi surrenali. I principali problemi connessi con vitiligine sono cosmetici, specialmente quando questa condizione si verifica in individui con pelle molto scura. A volte questa condizione va via senza trattamento. Per informazioni sui servizi di assistenza e informazioni sulla vitiligine vedere la risorsa seguente:
Storia
Caratteristiche morfologiche tipiche della sindrome di Waardenburg possono essere riconosciuti immediatamente o subito dopo la nascita. Caratteristiche includono tipicamente ciuffo bianco, ampio radice del naso, e iridi ipopigmentate.
I genitori notano che il bambino non reagisce ai suoni.
Esame Fisico
Non tutti i casi esprime tutte le manifestazioni cliniche della sindrome Waardenburg completa e le condizioni di forme fruste sono comunemente osservati. Sulla base di criteri clinici e genetici, sono riconosciute 4 tipi di sindrome di Waardenburg. Tutte le forme mostrano una marcata variabilità, anche all’interno delle famiglie.
|
|
Contrassegnata asimmetria facciale, lagoftalmo, un angolo cadenti destra della foce. Immagine per gentile concessione di Dourmishev LA et al, Cutis 1999 |
|
|
Il viso nel profilo dimostra mancanza dell’ angolo nasofrontale, ipertricosi delle sopracciglia, punta del naso all’insù, e labbro superiore accorciato con l’arco di Cupido pronunciato. Immagine per gentile concessione di Dourmishev LA |
|
|
Fratello e sorella con sindrome di Waardenburg. Immagine per gentile concessione di Dourmishev LA et al, Cutis 1999; 63:139-40 |



Tipo I Waardenburg
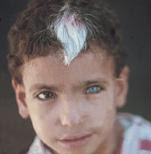 Il Tipo I Waardenburg è il tipo più comune ed è causata da una mutazione nel gene PAX3. Il gene PAX3 è parte di una famiglia di geni che è importante per lo sviluppo di tessuti e organi. Questo gene produce una proteina che controlla altri geni responsabili dello sviluppo di cellule specifiche e parti del corpo, quali ossa facciali e melanociti (cellule che compongono il pigmento melanina) manifestazioni cliniche (ad esempio, ciuffo bianco, macchie della pelle) sono più frequenti nel I tipo.
Il Tipo I Waardenburg è il tipo più comune ed è causata da una mutazione nel gene PAX3. Il gene PAX3 è parte di una famiglia di geni che è importante per lo sviluppo di tessuti e organi. Questo gene produce una proteina che controlla altri geni responsabili dello sviluppo di cellule specifiche e parti del corpo, quali ossa facciali e melanociti (cellule che compongono il pigmento melanina) manifestazioni cliniche (ad esempio, ciuffo bianco, macchie della pelle) sono più frequenti nel I tipo.
La melanina è responsabile per il colore del nostro occhi , capelli e pelle. Sono necessari anche melanociti nel nell’orecchio interno per l’udito. Pertanto, una mutazione nel gene PAX3 significa che le cellule pigmento melanina non possono svilupparsi adeguatamente, portando a vari problemi. Questi problemi comprendono chiazze di capelli bianchi, e le patch o aree di pelle che mancano pigmentazione. Gli occhi possono anche avere pigmentazione irregolare. Mancanza di melanina contribuisce anche alla neurosensoriale sordità associata alla sindrome. L’importo della perdita dell’udito può essere ovunque da moderata a profonda.
Il Tipo I Waardenburg è generalmente autosomica dominante – è ereditato da un solo genitore.
Tipo II Waardenburg
Waardenburg tipo II, anche un tipo più comune, perdita dell’udito neurosensoriale (77%) e eterocromia iride (47%) sono i due più importanti indicatori diagnostici di questo tipo. si possono anche avere cambiamenti di colore nei capelli, pelle . La differenza principale tra tipo I e tipo II è che nel tipo II, gli occhi non sono distanziati . Mentre si sospetta che questo tipo sia causato da mutazioni nel gene PAX3, si è anche creduto sia causata da mutazioni nel MITF (fattore di trascrizione associato a microftalmia-) ed ai geni SNAI2. Tipo II può essere ereditata sia nella modalità autosomica dominante (un genitore) che con modalità autosomica recessiva, il che significa che due genitori devono avere il gene per essere in grado di produrre un bambino con Waardenburg di tipo II.
Nei tipi autosomici dominanti, un bambino deve ricevere solo il gene WS da un genitore per avere WS. Nei tipi autosomici recessivi, un bambino deve ricevere il gene WS da entrambi i genitori per avere WS.
In aggiunta, ci sono cinque sottotipi di tipo II Waardenburg. Questi sottotipi sono Tipo IIA, che è causata da una mutazione nel gene MITF sul cromosoma 3; Tipo IIB, che è associato con cromosoma 1; Tipo IIC e IID, che coinvolgono il cromosoma 8; e Digitare IIE, una mutazione sul cromosoma 22.
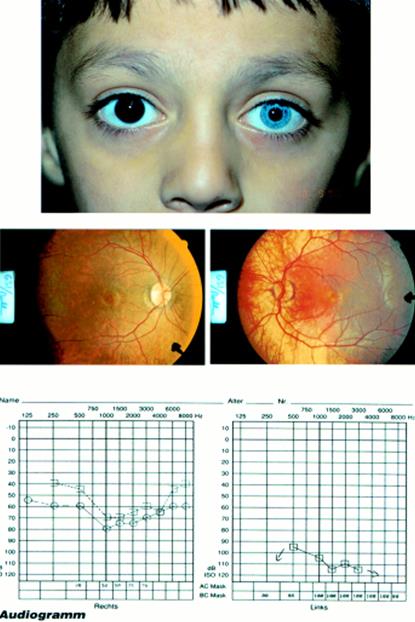
Sindrome di Waardenburg tipo 2 in una famiglia turca: implicazioni per l’importanza del modello di fondo pigmentazione
Tipo III Waardenburg
Poi c’è il Tipo III, un tipo molto raro. è più rara rispetto alle WS1 e WS2, ma è la
forma più grave. Può essere considerata come un sottotipo della WS1 dal momento che anch’essa è dovuta a mutazioni nel gene PAX3 (delezione eterozigote); di solito è sporadica e solo occasionalmente sono stati segnalati casi familiari ( Ciò che distingue il tipo III (anche causata da una mutazione nel gene PAX3) dagli altri è che nel Tipo III, esistono anomalie degli arti superiori, quali malformazioni delle braccia e delle mani, ma è anche caratterizzato da anomalie muscoloscheletriche (cioè, aplasia delle prime due costole, scapola alata, rigidità delle articolazioni, contratture articolari in flessione delle dita delle mani e dei piedi la mancanza di differenziazione delle piccole ossa carpali, la formazione cistica del sacro, anomalie del armi [ad esempio, amiloplasia e rigidità delle articolazioni). Ad esempio, le dita possono essere fuse insieme sindattilia cutanea bilaterale. Questo tipo di Waardenburg è anche conosciuta come sindrome di Klein-Waardenburg. Il Tipo III può sorgere spontaneamente (nessun legame genetico, si ha solo una mutazione ) oppure può essere ereditata da un genitore. o Altre manifestazioni cliniche della sindrome tipo III comprendono la piena sintomatologia della malattia, più ritardo mentale, microcefalia, e gravi anomalie scheletriche.
Figura 1: (a) Sindattilia delle dita medio e anulare delle mani e le dita dei piedi 2 ° e 3 ° di metri lungo con un forte emarginati, macule depigmentate sulla tibia destra. (B) Sindattilia delle dita dei piedi 2 ° e 3 ° del piede sinistro. (C) Sindattilia delle dita dei piedi 2 ° e 3 ° dei piedi giusti. (D) Close-up che mostra 2 ° e 3 ° fuso dita dei piedi
La WS3 è più spesso associata a perdita progressiva dell’udito neurosensoriale.
In un caso insolito, specialisti in India hanno diagnosticato una Waardenburg di tipo III in un bambino di 7 anni in cui si erano fusi il dito medio ed anulare su entrambe le mani, più due dita fuse. Una gamba aveva un cerotto bianco. I suoi occhi erano di colori diversi. Aveva una grande radice del naso (la zona tra gli occhi) e qualche capello bianco. Il padre del bambino era normale e non aveva sintomi. Ma la madre ha avuto sintomi, tra cui la sordità. In seguito è stata diagnosticata come tipo II.
Non è possibile capire la differenza tra WS di Tipo 1 e WS di Tipo 3 basandosi solo sull’aspetto fisico. I test genetici sono l’unico metodo accurato per distinguere tra i due tipi.
Tipo IV Waardenburg
L’ultimo tipo di sindrome di Waardenburg, anche rare, è di tipo IV. il tipo IV è un tipo II con malattia di Hirschsprung. Attualmente, sono stati identificati sei geni, onnicomprensivi nella Tabella 1. Questo tipo è anche conosciuta come sindrome di Waardenburg-Shah (associazione di sindrome di Waardenburg con la malattia di Hirschsprung).Il tipo 4 è raro, con solo 48 casi segnalati fino al 20026 Egbalian F (2008).
Oltre alle caratteristiche tipiche di Waardenburg, questo modulo comprende anche la malattia di Hirschsprung, un problema digestivo che causa stitichezza o blocco intestinale. Il tipo IV viene solitamente tramandato da un genitore, ma può anche essere ereditata da entrambi i genitori.
Le persone con WS4 possono anche avere una condizione chiamata malattia di Hirschsprung (HD). HD di solito viene scoperto poco dopo la nascita, e può essere in pericolo di vita. È una condizione che comporta problemi del colon e l’incapacità di espellere feci dall’intestino. Qualcuno con HD non trattati sviluppa un colon allargata, stipsi cronica, e ritardo di crescita. HD è trattata con la chirurgia correttiva o modificazione della dieta.
Il tipo IV dispone anche di sottotipi, ognuno causata da una mutazione del gene diverso.
La sindrome di Waardenburg-Shah (WSS) è una neurocristopatia caratterizzata dall’associazione tra la sindrome di Waardenburg (sordità neurosensoriale e anomalie della pigmentazione; si veda questo termine) e la malattia di Hirschsprung (si veda questo termine). La prevalenza è sconosciuta ma sono stati riportati oltre 50 casi. I pazienti, nel periodo neonatale, presentano anomalie della pigmentazione (frezza sulla fronte, sopracciglia e ciglia bianche, eterocromia dell’iride, macchie cutanee bianche) in associazione a ostruzione intestinale. La sordità neurosensoriale è comune, precoce e può essere unilaterale. Lo sviluppo psicomotorio è normale. La WSS è causata da anomalie della migrazione/differenziazione delle cellule della cresta neurale durante lo sviluppo embrionale. Fino ad ora sono stati identificati tre geni-malattia: il gene EDNRB (13q22.3), che codifica il recettore dell’endotelina-B, il gene EDN3 (20q13.32), che codifica il ligando del recettore dell’endotelina e il gene SOX10 (22q13.1), che codifica il fattore di trascrizione SOX10. Le mutazioni dei geni EDNRB e EDN3 si trasmettono con modalità autosomica recessiva; i soggetti omozigoti per le mutazioni in questi geni presentano la WSS, mentre quelli eterozigoti presentano la malattia di Hirschsprung isolata o, spesso, sono asintomatici. Le mutazioni del gene SOX10 vengono ereditate con modalità autosomica dominante e alcune specifiche mutazioni (in particolare quelle che interessano i due ultimi esoni codificanti) si associano a una forma più grave della WSS, con quadri neurologici caratterizzati da neuropatia periferica demielinizzante, leucodistrofia centrale dismielinizzante, sindrome di Waardenburg e malattia di Hirschsprung (PCWH; si veda questo termine). Viceversa, alcuni soggetti con la sindrome di Waardenburg tipo 2 (si veda questo termine) sono portatori delle mutazioni eterozigoti del gene SOX10. Non sono state riscontrate mutazioni nei tre geni-malattia nel 20-40% dei soggetti affetti da WSS/PCWH. La diagnosi si basa sul riconoscimento del quadro clinico e viene confermata dal riscontro di mutazioni in uno dei geni-malattia. La diagnosi differenziale si pone con altre forme della sindrome di Waardenburg (1, 2 e 3; si vedano questi termini) e anche con altre malattie rare, come il piebaldismo e le sindromi ABCD o BADS (sempre associate a mutazioni del gene EDNRB; si vedano questi termini). Alle famiglie nelle quali viene riscontrata la mutazione patogenetica deve essere offerta la diagnosi prenatale molecolare. La consulenza genetica deve adattarsi alla modalità di trasmissione della mutazione identificata. Il trattamento è solo sintomatico; il trattamento della malattia di Hirschsprung deve comprendere anche la chirurgia e la sordità deve essere trattata. La prognosi è spesso buona, ma la sindrome può associarsi a elevata morbilità e mortalità, secondarie alle complicazioni della malattia di Hirschsprung (legate alle dimensioni del segmento di intestino aganglionico).
Diagnosi
I bambini nati con la sindrome di Waardenburg possono avere i cambiamenti caratteristici dei capelli e della pelle e perdita dell’udito. Tuttavia, se i sintomi sono lievi, la sindrome Waardenburg può non essere diagnosticata fino a quando non viene diagnosticata un membro della famiglia ed allora tutti i membri della famiglia sono esaminati. Test dell’udito formali possono essere utilizzati per rilevare la perdita dell’udito.
Trattamento
Diversi i sintomi della sindrome di Waardenburg appaiono in persone diverse, anche all’interno della stessa famiglia. Alcuni individui non richiedono alcun trattamento, mentre altri possono avere bisogno di un intervento chirurgico o degli occhi o altre anomalie. Non sono necessari dieta o attività particolari restrizioni, e la sindrome di Waardenburg di solito non influenzano la mente.
La consulenza genetica
A causa del modo con cui la sindrome di Waardenburg (autosomica dominante) di tipo 1 e 2 è ereditata, un individuo affetto ha una probabilità del 50%, ad ogni gravidanza, di avere un figlio affetto. Dal momento che i sintomi possono variare, non c’è modo di prevedere se un bambino affetto avrà sintomi più lievi o più gravi rispetto la sua / il suo genitore. Ereditarietà dei tipi 3 e 4 è più complessa, ma la consulenza genetica può aiutare a valutare il rischio di trasmettere la sindrome di Waardenburg a un bambino.
Diagnosi differenziale
· Piebaldismo
· Sindrome di Vogt-Koyanagi-Harada
Studi di laboratorio
Tipi di sindrome di Waardenburg 1 e 3 sono più comunemente associati con mutazioni puntiformi in PAX3, e di tipo 2 è associato con MITF mutazioni puntiformi. Il piebaldismo (OMIM 17200) è una rara patologia autosomico dominante caratterizzata dalla presenza di macchie ipopigmentate
a livello della parte mediana della fronte e diffuse a livello di torace, addome ed arti. È causato da mutazioni
inattivanti o delezioni del gene KIT o del gene SLUG.Legatura-dipendente multipla della sonda amplificazione può essere utilizzato per rilevare cambiamenti nei geni bersaglio Milunsky JM,et al.,2007.
Altri test
Partington ha usato le 3 distanze interoculari per determinare la presenza di distopia cantorum. (Il valore referente tra distanza angoli palpebrali mediale delle palpebre e la lunghezza tra gli alunni era di 0,6 mm.)
· Tra gli angoli cantali interni
· La vicina distanza papillare
· Tra gli altri angoli cantali
Arias e Mota ha sviluppato l’indice W per la diagnosi di spostamento laterale della angoli palpebrali interiore. (Indice AW superiore a 2,07 mostra dystopy, mentre un indice inferiore a 1,87 è normale.)
· W index = X + Y + alb
· Dove X = [a – {0.21119c + 3,909}] / c e Y = [2a – {0.2497b + 3,909}] / b in mm
· Distanza a è tra angoli palpebrali interni; distanza b, tra gli alunni; e la distanza c, tra angoli palpebrali esterno.
I risultati istologici
Gli studi istochimici nella pelle acromica delle persone con sindrome di Waardenburg mostrano che i melanociti sono assenti, o che solo poche cellule diidrossifenilalanina-positive sono presenti.
Osservazioni ultrastrutturali non rivelano melanociti, cellule dendritiche indeterminati, o melanosomi nei cheratinociti della pelle depigmentazione; tuttavia, il numero di cellule di Langerhans nell’epidermide è normale. Il numero di melanociti sul bordo delle leucodermatiti è ridotta, e numerose anomalie nucleari citoplasmatici sono noti. Alcuni melanosomi circondate da un alone chiaro si trovano all’interno dei vacuoli. Il numero dei melanociti nelle zone ipopigmentate della sindrome di Waardenburg è drasticamente ridotto. Queste cellule dendritiche contengono melanosomi mal melanizzate.
Esame istopatologico delle orecchie interne delle persone con sindrome di Waardenburg mostra organi assenti Corti, atrofia del ganglio spinale, e una riduzione del numero di fibre nervose.
Tipi di WS 1 e 3
Tipi di WS 1 e 3 hanno caratteristiche fisiche molto simili. Tipi 1 e 3 hanno un modello di ereditarietà autosomica dominante. Questo significa che se un genitore ha il gene per WS e l’altro genitore non lo fa, i loro figli hanno una probabilità del 50% di avere WS.
WS1 è associato con congeniti, variabili neurosensoriali (permanenti) perdite uditive.
WS3 è più spesso associata a perdita progressiva dell’udito neurosensoriale.
Entrambi i tipi hanno le caratteristiche fisiche descritte altrove su questo sito. Queste funzionalità includono eterocromia, luminosi occhi azzurri, distopia dei canti, e le anomalie del pigmento.
WS3 ha le caratteristiche aggiuntive di anomalie degli arti superiori e minori contratture articolari delle dita delle mani e dei piedi.
Non è possibile capire la differenza tra WS Tipo 1 e Tipo 3 WS basata sull’aspetto fisico. Test genetici è l’unico modo accurato per distinguere tra i due tipi.
Gli individui con WS2 possono avere caratteristiche simili a quelle di WS 1 e 3, tranne che dei canti distopia e anomalie muscoloscheletriche non sono presenti.
WS di tipo 2
-
- WS2 ha diversi sottotipi in base alla posizione del gene specifico che causa problemi. La maggior parte dei sottotipi sono autosomica dominante in eredità, ma alcuni sono autosomica recessiva. Nei tipi autosomiche dominanti, un bambino deve ricevere solo il gene WS da un genitore per avere WS. Nei tipi autosomiche recessive, un bambino deve ricevere il gene WS da entrambi i genitori per avere WS.
- Le principali caratteristiche del WS2 sono iridis heterochromia e ipoacusia neurosensoriale.
WS Type 4 è anche chiamato sindrome di Shah-Waardenburg.
- Persone con WS4 possono avere uno o tutti i sintomi di WS2.
Le persone con WS4 hanno una condizione chiamata malattia di Hirschsprung (HD). HD di solito viene scoperto poco dopo la nascita, e può essere in pericolo di vita. HD è una condizione che comporta problemi al colon, e l’incapacità di espellere feci dall’intestino. Qualcuno con HD svilupperà un colon allargata, la stipsi cronica, ritardo di crescita, e, se l’intervento chirurgico è riuscito, può morire.
I sottotipi della sindrome sono riconducibili a diverse varianti genetiche:
|
Tipo |
Gene |
Conosciuto anche come |
||
|
Tipo I, WS1 |
2q35 |
– |
||
|
Tipo IIa, WS2A (originariamente WS2) |
3p14.1-p12.3 |
– |
||
|
Tipo IIb, WS2B |
1p21-p13.3 |
– |
||
|
Digitare IIc, WS2C |
8p23 |
– |
||
|
Digitare IId, WS2D (molto raro) |
8q11 |
– |
||
|
Tipo III, WS3 |
2q35 |
Sindrome di Klein-Waardenburg |
||
|
Tipo IVa, WS4A |
13q22 |
|||
|
Tipo IVb, WS4B |
20q13 |
|||
|
Digitare IVc, WS4C |
22q13 |
|
Altri collaboratori
-
- Sindrome di Waardenburg-Klein prende il nome Petrus Johannes Waardenburg (1886-1979), un oculista olandese e genetista, e David Klein , un genetista umano svizzero e oculista.
-
- La sindrome di Mende II è il nome di Irmgard Mende (1938 -), un medico tedesco-americano.
-
- Van der Sindrome Hoeve-Halbertsma-Waardenburg prende il nome da Jan Van der Hoeve (1878-1952), un oculista olandese, Nicolaas Adolf Halbertsma (1889-1968), medico olandese e Petrus Johannes Waardenburg (1886-1979).
-
- Van der sindrome Hoeve-Halbertsma-Gualdi prende il nome da Jan Van der Hoeve (1878-1952), Nicolaas Adolf Halbertsma (1889-1968) e Vincenzo Gualdi (1891-1976), un medico italiano.
- La sindrome di Vogt è chiamato per Cecile Vogt (1875-1962), un neuropatologa franco-tedesca.
BIBLIOGRAFIA
Arias, S., & Mota, M. (1978). Apparent non-penetrance for dystopia in Waardenburg’s syndrome type 1, with some hints on the diagnosis of dystopia canthorum. J. Genet. Hum., 26 , 103-131.
^ Arias S (1971). “Genetic Heterogeneity in the Waardenburg Syndrome”. Birth Defects Original Article Series 7 (4): 87–101.
Baar-Hamilton, R., Matheson, L., & Keay, D. (1991). Ototoxicity of cis-platinum and its relationship to eye colour. The Journal of Laryntology and Otology, 105, 7-11.
Baldwin, C., Hoth, C., Macina, R., & Milunsky, A. (1995). Mutation in PAX3 that causes Waardenburg’s syndrome type I: Ten new mutations and review of the literature. American Journal of Medical Genetics, 58 : 115-122.
Bandyopadhyay, S., Prasad, S., & Singhania, P. (1999). Partial anodontia in a case of Waardenburg’s syndrome. The Journal of Laryngology and Otology, 113 : 672-674.
^ Baral, Viviane, et al. “Screening of MTIF and SOX10 Regulatory Regions In Waardenburg Syndrome Type 2. “Plos ONE 7.7 (2012): 1-8. Academic Search Premier. Web. 4 Apr. 2014.
Black, F., Pesznecker, S., Allen, K., & Gianna, C. (2001). A vestibular phenotype for Waardenburg syndrome. Otology & Neurotology, 22 : 188-194.
Bondurand N, Pingault V, Goerich DE, et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet. Aug 12 2000;9(13):1907-17. [Medline].
Cagatay, O., Baserer, N., & Tinaz, N. (2000). Audiometric manifestations of Waardenburg’s syndrome. Ear, Nose & Throat Journal.
Chan KK, Wong CK, Lui VC, Tam PK, Sham MH. Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: Differential effects on target gene regulation. J Cell Biochem. Oct 15 2003;90(3):573-85. [Medline].
Chang T, Hashimoto K, Bawle EV. Spontaneous contraction of leukodermic patches in Waardenburg syndrome. J Dermatol. Nov 1993;20(11):707-11. [Medline].
Da-Silva, E., Batista, J., Medeiros, M., & Fonteless, S. (1993). Craniofacial anthropometric studies in Waardenburg’s syndrome type I. Clinical Genetics, 44 : 20-25.
DeStefano AL, Cupples LA, Arnos KS, et al. Correlation between Waardenburg syndrome phenotype and genotype in a population of individuals with identified PAX3 mutations. Hum Genet. May 1998;102(5):499-506. [Medline].
Dennis, L., Lanza, J., & Har-el, G. (1996). Waardenburg Syndrome. Otoltolaryngol Head Neck Surgery, 114 : 166-167.
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. Sep 1999;38(9):656-63. [Medline].
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg’s syndrome with facial palsy and lingua plicata: is that a new type of disease?. Cutis. Mar 1999;63(3):139-41. [Medline].
Dundar M, Lowther G, Colgan J, et al. A case with Waardenburg syndrome presenting with two separate translocations–one reciprocal and one complex. Clin Dysmorphol. Jan 2001;10(1):65-6. [Medline].
^ Egbalian F (2008). “Waardenburg-Shah Syndrome; A Case Report and Review of the Literature” . Iranian Journal of Pediatrics 18(1): 71–74.
Fisch, L. (1959). Deafness as part of an hereditary syndrome. The Journal of Laryntology and Otology , 355-382.
Galasso C, Bombardieri R, Cerminara C, Stranci G, Curatolo P. Anophthalmia-Waardenburg syndrome with expanding phenotype: does neural crest play a role?. J Child Neurol. Nov 2007;22(11):1252-5.[Medline].
^ Geigy CA, Heid S, Steffen F, Danielson K, Jaggy A, Gaillard C (2007). “Does a pleiotropic gene explain deafness and blue irises in white cats?”. Veterinary Journal 173 (3): 548–553.
Graned, G., Collet, L., & Morgon, A. (1995). Physiopathological investigations in a family with a history of Unilateral hereditary deafness. Acta Otolaryngology (Stockh), 115 : 196-201.
Gupta, V., Harisch M., & Aggarwal, C. (2000). Open angle Glaucoma as a manifestation of Waardenburg’s syndrome. Indian J Opthalmol, 48 : 49-50.
Hildesheimer M., Maayan Z., Muchnik C., Rubinstein M., Goodman R.M. Auditory and vestibular findings in Waardenburg’s type II syndrome J. Laryngol. Otol. 1989 ; 103 : 1130-1133
Jung HJ, Jin SA, Choi SJ, Lee SC, Yun SJ. A de novo SOX10 mutation in apatient with Waardenburg syndrome type IV. J Am Acad Dermatol. 2013;68(6):[Medline].
Kadoi, C., Hayasaka, S., & Yamamoto, S. (1996). Branch retinal vein occlusion in a patient with Waardenburg Syndrome. Ophthalmologica, 210 : 354-357.
Keefe, M. (1999). A typical presentation of Waardenburg’s syndrome. Otolaryngol Head Neck Surgery, 120: 749.
Keats, J. (2002). Genes and syndromic hearing loss. Journal Of Communication, 35: 355-366.
Klein, D. (1983). Historical background and evidence for dominant inheritance of the Klein-Warrensburg syndrome(type III). American Journal of Medical Genetics, 14: 231-239.
^Klein-Waardenburg syndrome at Who Named It?
Knox, G., Mcpherson, A., & Lopes, A. (1999). A clinical presentation of Waardenburg’s syndrome type II .
^ Kumar, Sudesh, and Kiran Rao. “Waardenburg Syndrome: A Rare Genetic Disorder, A Report of Two Cases.” Indian Journal of Human Genetics 18.2 (2012): 254-255. Academic Search Premier. Web. 4 Apr. 2014.,
Lee, D., Lanza, J., & Har-el, G. (1996). Waardenburg Syndrome. Otolaryngol Head Neck Surgery , 114: 166-167.
Liu, X., Newton, E., & Read, A. (1995). Hearing loss and pigmentary disturbances in Waardenburg syndrome with reference to WS Type II . The Journal of Laryntology and Otology , 109: 96-100.
Liu, X., Newton, E., & Read, A. (1995). Waardenburg’s syndrome type II: Phenotype findings and diagnostic criteria. American Journal of Medical Genetics, 55: 95-100.
Liu, X. & Newton, E. (1997). Distortion product emissions in normal-hearing and low-frequency hearing loss carriers of genes for Waardenburg’s syndrome. Ann Otol Rhinol Larygology, 106: 220-225.
^ Matulich E. “Deafness in Ferrets” . Cypresskeep.com.
Mckenna, M., Milunsky, A., Baldwin, C., & Nadol, J. (2001). Otopathology in a case of type 1 Waardenburg’s syndrome. Ann Otol Rhinol Otolaryngology, 110.
Madden C, Halsted MJ, Hopkin RJ, Choo DI, Benton C, Greinwald JH Jr. Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope. Nov 2003;113(11):2035-41.[Medline].
^ Matulich E. “Deafness in Ferrets” . Cypresskeep.com.
Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MLPA in Waardenburg syndrome. Genet Test. Summer 2007;11(2):179-82. [Medline].
Mishriki, Y. (2000). Facial clues to an inherited syndrome. Postgraduate Medicine , 106.
Moore SW, Johnson AG. Hirschsprung’s disease: genetic and functional associations of Down’s and Waardenburg syndromes. Semin Pediatr Surg. Aug 1998;7(3):156-61. [Medline].
Morell, R., Friedman.,T., Asher, J., & Robbins, L. (1996). The incidence of deafness is non-randomly distributed among families segregating Waardenburg syndrome type 1 (WS1).
Morell R, Carey ML, Lalwani AK, Friedman TB, Asher JH Jr. Three mutations in the paired homeodomain of PAX3 that cause Waardenburg syndrome type 1. Hum Hered. Jan-Feb 1997;47(1):38-41. [Medline].
Nakashima, S., Sando, I., Takahashi, H., & Hashida, Y. (1992). Temporal bone histopathology findings of Waardenburg’s syndrome: A case report. Laryngoscope, 102: 563-567.
Newton, V. (1990). Hearing loss and Waardenburg’s syndrome: implications for genetic counseling. The Journal of Laryntology and Otology, 104, 97-103.
Ortonne, J. (1988). Piebaldism, Waardenburg’s syndrome, and related disorders. Dermatologic Clinics, 6, 205-216.
Oshimo T, Fukai K, Abe Y, Hozumi Y, Yokoi T, Tanaka A, et al. Pediatric case report: clinical profile of a patient with PCWH withp.Q377X nonsense mutation in the SOX10 gene. J Dermatol. 2012;39(12):1022-5.[Medline].
Oysu C, Baserer N, Tinaz M. Audiometric manifestations of Waardenburg’s syndrome. Ear Nose Throat J. Sep 2000;79(9):704-9. [Medline].
^Petrus Johannes Waardenburg at Who Named It?
Read, A. & Newton, V. (1997). Waardenburg syndrome. Journal of Medical Genetics, 34: 656-665.
Read, A. (2000). Waardenburg syndrome. Genetics in Otorhinolaryngology Adv Otorhinolaryngol, 56: 32-38.
Read, A. (2000). Hereditay deafness: lessons for developmental studies and genetic diagnosis. Eur J Pediatr, 159: 232-235.
Reed, W., Stone, V., Boder, E., & Ziprowski, L. (1967). Pigmentary disorders in association with congenital deafness. Arch Derm, 95, 176-178.
Reynolds, J., et al. (1995). Analysis of variability of clinical manifestations in Waardenburg syndrome. American Journal of Medical Genetics, 57: 540-547.
^ Richards J (1999). ASPCA Complete Guide to Cats: Everything You Need to Know About Choosing and Caring for Your Pet .Chronicle Books. p. 71.
Rosenberg, T., et al. (1987). Chroidereima, congenital deafness and mental retardation in a family with an X chromosomal deletion. Ophthalmic Paediatrics and Genetics, 8: 139-143.
Sayh, B., Akarsu, A., & Altan, S. (1995). Anophthalmos-Syndactyly(Waardenburg) syndrome without oligodactyly of toes. American Journal of Medical Genetics, 58: 18-20.
Sener, R. (1998). Cranial MR imaging findings in Waardenburg syndrome: exophthalmia, and hypothalamic hamartoma. Computerized Medical Imaging and Graphics, 22: 409-411.
Sheffer, R. & Zlotogora, J. (1992). Autosomal dominant inheritance of Klein Waardenburg’s syndrome. American Journal of Medical Genetics , 42: 320-322.
Shim WK, Derieg M, Powell BR, Hsia YE. Near-total intestinal aganglionosis in the Waardenburg-Shah syndrome. J Pediatr Surg. Dec 1999;34(12):1853-5. [Medline].
Smith, S., Kolodziej, P., & Olney, A. (1998). Waardenburg Syndrome. Ear, Nose& Throat Journal, 77:257-258.
Sotirova, V., et al. (1999). Identification of a novel mutation in the paired domain of PAX3 in an Iranian family with Waardenburg’s Sydrome type 1. Opthalmic Genetics , 21:25-28.
Strain, M. et al.(1992). Brainstem auditory-evoked potential assessment of congenital deafness in Dalmatians: Associations with phenotypic markers. Journal of Veterinary Internal Medicine , 6: 175-182.
Sudip Kumar Ghosh, Debabrata Bandyopadhyay, Arghyaprasun Ghosh, Surajit Kumar Biswas, Rajesh Kumar Mandal.Waardenburg syndrome: A report of three cases. Indian Journal of Dermatology, Venereology and Leprology. Vol. 76, Num. 5, 2010, pp. 550-552
Suyugul, Z., et al. (1996). Anophtalamia-Waardenburg Syndrome: A report of three cases. American Journal of Medical Genetics, 62: 391-397.
Sznajer Y, Coldea C, Meire F, Delpierre I, Sekhara T, Touraine RL. A de novo SOX10 mutation causing severe type 4 Waardenburg syndrome without Hirschsprung disease. Am J Med Genet A. Apr 15 2008;146A(8):1038-41. [Medline].
Tachibana M. Evidence to suggest that expression of MITF induces melanocyte differentiation and haploinsufficiency of MITF causes Waardenburg syndrome type 2A. Pigment Cell Res. Feb-Apr 1997;10(1-2):25-33. [Medline].
Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol. Jul-Aug 2006;72(4):326. [Medline].
Tekin M, Bodurtha JN, Nance WE, et al. Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3:a simple variant or a true syndrome? Clinical Genetics 2001;60:301-4
Valenzuela, G., et al. (1995). Waardenburg syndrome and Gastric Stasis in adults.
Verheij JB, Sival DA, van der Hoeven JH, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur J Paediatr Neurol. Jan 2006;10(1):11-7.[Medline].
^ Yang, T, et al. “Double Heterozygous Mutations Of MTIF and PAX3 Result in Waardenburg Syndrome With Increased Penetrance In Pigmentary defects.” Clinical Genetics 83.1 (2013): 78-62. Academic Search Premier. Web. 3 Apr. 2014.
Yoder, B. & Preyson, R. (2002). Shah-Waardenburg syhndome and dandy-walker formation: an autopsy report. Clinical Neuropathology , 21: 236-240.
^ Jump up to:ab Waardenburg PJ (September 1951). “A New Syndrome Combining Developmental Anomalies of the Eyelids, Eyebrows and Noseroot with Pigmentary Anomalies of the Iris and Head Hair and with Congenital Deafness; Dystopia canthi medialis et punctorum lacrimalium lateroversa, Hyperplasia supercilii medialis et radicis nasi, Heterochromia iridum totalis sive partialis, Albinismus circumscriptus (leucismus, poliosis) et Surditas congenita (surdimutitas) “ . Am. J. Hum. Genet. 3 (3): 195–253.
^ Webb AA, Cullen CC (2010). “Coat color and coat color pattern-related neurologic and neuro-ophthalmic diseases” . The Canadian Veterinary Journal 51 (6): 653–657.
Wildhardt G, Zirn B, Graul-Neumann LM, Wechtenbruch J, Suckfüll M, Buske A, et al. Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics.BMJ Open. 2013;18;3(3):[Medline].
Zhang H, Luo H, Chen H, Mei L, He C, Jiang L, et al. Functionalanalysis of MITF gene mutations associated with Waardenburg syndrome type 2. FEBS. 30;586(23);2012:4126-31. [Medline].
Books
Cremers, WRJ & Smith, RJH (2002). Genetic hearing impairment: It’s clinical presentations (Advances in oto-rhino-laryngology) . New York, NY: S.Karger AG. (ISBN# 3085574495)
Jones, KL & Smith, DW (2006). Smith’s recognizable patterns of human malformation . Philadelphia, PA: Elsevier, Inc.(ISBN# 0721661157)
Kahn, A. (2007). Waardenburg Syndrome. San Diego, CA: Plural Publishing, Inc. (ISBN# 1597560219)
Toriello, HV, Reardon, W. & Gorlin, RJ (2004). Hereditary hearing loss and its syndromes (Oxford monographs on medical genetics) . New York, NY: Oxford University Press, Inc.(ISBN# 019513849X)
Wiedemann, HR & Kunze, J. (1997). Clinical syndromes (3rd ed.) . Italy: Mosby-Wolfe.(ISBN# 0723429502)
Willems, PJ (2004). Genetic hearing loss . New York, NY: Marcel Dekker, Inc. (ISBN# 0824743091)
Sindrome branchiootorenale BOR, Sindrome BOR, Displasia branchiootorenale, Branchio-Otorenale Displasia,Branchio-Otorenale Syndrome, Sindrome Branchio-Oto-Renale, Sindrome di Melnick-Fraser
– trasmissione autosomica dominante Fig.1
– espressività variabile
– sordità neurosensoriale. mista, trasmissiva Fig.2
– boffoni preauricolari Fig.3
– cisti-fistole del collo (branchiali) Fig.4
– malformazione del padiglione auricolare Fig.5
– malformazioni renali (agenesia-ipoplasia)Fig.6
|
|
|
|
|
Fig.1
|
Fig.2 |
Fig. 3 |
|
|
|
|
|
Fig.4 |
Fig.5 |
Fig.6 |

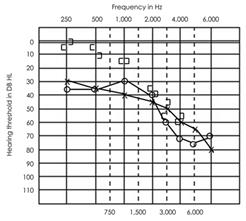
Che cosa è la sindrome Branchio-Oto-Renale?,pag.2
Quali geni sono legati alla sindrome Branchio-Oto-Renale?,pag.3
Come i pazienti ereditano la sindrome Branchio-Oto-Renale ?,pag.3
Manifestazioni otorinolaringoiatriche, ?pag.3
Eziologia e la patogenesi, pag.4
Diagnosi, Criteri principali, Criteri minori,pag.6
APPROFONDIMENTI
Famiglia con sindrome Branchio-Oto-Renale: correlazioni cliniche e genetiche,pag.11
Un caso di Sindrome di Branchio-Oto-Renale,pag.18
La sindrome Branchio-oculo-facciale e le sindromi branchio-otic/branchio-oto-renale sono entità distinte,pag.22
Che cosa è la sindrome Branchio-Oto-Renale?
LA Sindrome Branchio-Oto-Renale (BOR) è una condizione genetica, nota anche come sindrome di Melnick-Fraser, è caratterizzata da un’associazione di: 1) fistole brachiale o cisti lungo i lati del collo corrispondenti alla posizione delle fessure branchiali embriologiche; 2) malformazioni dell’orecchio, che possono includere l’orecchio interno, medio ed esterno; 3) malformazioni renali, che possono variare in gravità da ipoplasia renale alla agenesia con conseguente insufficienza renale. I segni ed i sintomi di questa condizione , tuttavia, possono variare.
“Branchio-” si riferisce al secondo arco branchiale, che è una struttura che negli embrioni dà origine ai tessuti nella parte anteriore e laterale del collo. Nelle persone con sindrome branchiootorenale , sviluppo anormale del secondo arco branchiale possono causare la formazione delle masse al collo chiamato cisti branchiale leporino. In alcune persone, connessioni anormali chiamati modulo fistole passaggi tra queste cisti e la superficie del collo. Fistole può anche svilupparsi tra la pelle del collo e della gola, vicino alle tonsille. Cisti schisi branchiali e fistole possono causare problemi di salute se diventano infette.
“Oto-” si riferisce all’orecchio; la maggior parte delle persone con sindrome branchiootorenale hanno perdite e altre anomalie dell’orecchio dell’udito. La perdita dell’udito è conosciuta come la sordità neurosensoriale se è causato da cambiamenti nella parte interna dell’orecchio e sordità conduttiva se è causato da cambiamenti dell’orecchio medio. La sindrome Branchio-Oto-Renale può anche comportare la perdita che deriva da cambiamenti sia l’orecchio interno e l’orecchio medio, che si chiama perdita uditiva mista dell’udito. Altre anomalie dell’orecchio associate alla sindrome di Branchio-Oto-Renale comprendono malformazioni dell’orecchio interno o l’orecchio medio e orecchio esterno di forma anomala (padiglioni auricolari). Alcune persone colpite hanno anche piccoli fori nella pelle (pit preauricolari) o piccoli lembi di pelle (tag preauricolari) proprio di fronte l’orecchio.
“Renale” si riferisce ai reni; La sindrome Branchio-Oto-Renale provoca alterazioni della struttura e della funzione renale. Queste anomalie vanno da lievi a gravi e possono interessare uno o entrambi i reni. In alcuni casi, la malattia renale allo stadio terminale (ESRD) si sviluppa più tardi nella vita. Questa grave condizione si verifica quando i reni diventano incapaci di filtrare efficacemente i liquidi ei prodotti di scarto dal corpo.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Ereditata in modo autosomico dominante, ogni bambino di un genitore con la sindrome di BOR ha una probabilità del 50% di presentare con la malattia. Si pensa che il 90% dei casi di sindrome BOR sono dovuti a eredità, mentre il rimanente 10% dei casi sono dovuti a mutazioni acquisite. BOR mostra espressività variabile, che rappresentano le differenze nella gravità dei sintomi tra i membri della famiglia. Penetranza, tuttavia, è 100%. Attesa (propensione per le generazioni successive di presentare con la malattia in età più giovane) non si verifica.
Storia
• Nel 1864 Heusinger è stato il primo a riconoscere un’associazione tra branchiale fistole leporino, pozzi preauricolari, e compromissione dell’udito.
• Nel 1975, Melnick et al. e Fraser et al. descritto sindrome BOR come entità specifica con un pattern di ereditarietà autosomica dominante.
Epidemiologia, Quanto è diffusa la sindrome Branchio-Oto-Renale ?
I ricercatori stimano che la sindrome Branchio-Oto-Renale colpisce circa 1 su 40.000 persone.
Quali geni sono legati alla sindrome Branchio-Oto-Renale ?
Le mutazioni in tre geni, EYA1, SIX1 e SIX5, sono noti per causare la sindrome Branchio-Oto-Renale . Circa il 40 per cento delle persone con questa condizione hanno una mutazione nel gene EYA1. Eredità autosomica dominante indica che il gene difettoso responsabile di una malattia si trova in una autosoma , e solo una copia del gene è sufficiente a causare la malattia, quando ereditato da un genitore che ha il disturbo. Mutazioni SIX1 eSIX5 sono cause molto meno comuni della malattia.
Le proteine prodotte dal EYA1, SIX1, ei geni SIX5 svolgono un ruolo importante nello sviluppo prima della nascita. La proteina EYA1 interagisce con diverse altre proteine, tra cui SIX1 e SIX5, per regolare l’attività dei geni coinvolti in molti aspetti dello sviluppo embrionale. La ricerca suggerisce che queste interazioni proteine sono essenziali per la normale formazione di molti organi e tessuti, tra cui il secondo arco branchiale, orecchie e reni. Mutazioni nel EYA1, SIX1, o gene SIX5 spesso interrompono la capacità delle proteine ‘di interagire uno con l’altro e regolare l’attività dei geni. Le conseguenti variazioni genetiche influenzano lo sviluppo di organi e tessuti prima della nascita, che porta gli elementi caratteristici della sindrome Branchio-Oto-Renale .
Mutazioni Eya1 e SIX1 sono stati trovati anche in alcune persone con una condizione nota come branchio-oto (BO) sindrome. Questa condizione include molte delle stesse caratteristiche come la sindrome branchiootorenale, ma individui affetti non hanno renali (renale) anomalie. Le due condizioni sono altrimenti così simili che i ricercatori spesso li considerano insieme (BOR / sindrome BO). Non è chiaro il motivo per cui alcune mutazioni Eya1 e SIX1 sono associati ad anomalie renali e altri non lo sono.
Alcune persone con la sindrome di Branchio-Oto-Renale non hanno una mutazione identificata nel EYA1, SIX1,o gene SIX5. In questi casi, la causa della condizione è sconosciuta.
Per saperne di più sul EYA1 , SIX1 , e SIX5 geni.
Come i pazienti ereditano la sindrome Branchio-Oto-Renale ?
Questa condizione è ereditata come carattere autosomico dominante, il che significa una sola copia del gene alterato in ogni cella è sufficiente a causare il disturbo. In circa il 90 per cento dei casi, una persona affetta eredita la mutazione da un genitore affetto. I casi rimanenti derivano da nuove mutazioni nel gene, e si verificano in persone senza storia di malattia nella loro famiglia.
Manifestazioni otorinolaringoiatriche
Le malformazioni auricolari comprendono pozzi preauricolari , che sono ciechi depressioni capocchia di spillo nella parte superiore del padiglione auricolare dell’orecchio. Essi si osservano in 75-85% dei pazienti. Altre malformazioni dell’orecchio esterno (40-60%) possono includere tag preauricolari, lop o pipistrello orecchie e microtia. I canali uditivi esterni possono essere atresici.
Anomalie dell’orecchio medio comprendono anomalie del ossicini, il nervo facciale, ei canali di Falloppio. La tomografia computerizzata delle ossa temporali può dimostrare coclea ipoplasico e canali semicircolari assenti o ipoplasia. È stata inoltre segnalata Mondini displasia. La perdita dell’udito (75-90%) di solito è stabile e può essere conduttiva, neurosensoriale o mista. Si stima che il 2% dei bambini con sordità profonda hanno la sindrome BOR.
Descritta per la prima nel 1975, da M. Melnick, e nel 1978 da FC Fraser. La sindrome di BOR (nota anche come sindrome di Melnick-Fraser) è l’associazione di malformazioni auricolari, fistole branchiali(63%) della sindrome BOR sono solitamente bilaterale e nella parte inferiore del collo con aperture esterne sul bordo mediale del muscolo sternocleidomastoideo, sordità e anomalie renali. La prevalenza è stimata in 1 caso ogni 40.000 popolazione generale.tole branchiali. Altre manifestazioni associate comprendono l’aplasia o stenosi dei dotti lacrimali (8-9%), palato alto arcuato o spacco, morso profondo, e anomalie del nervo facciale. Alcuni autori considerano la microsomia emifacciale (HFM) per essere una forma grave. Vedere emifacciale microsomia, nella sezione Sindrome di Goldenhar. La sindrome branchio-oto-renale (BOR) associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000(Fraser F.C.). Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un’interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un’ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell’orecchio esterno (malformazioni del padiglione, aplasia dell’orecchio, encondromi, stenosi dei condotti uditivi) (Fig. 4 ), dell’orecchio medio (esiste una componente di trasmissione all’audiogramma) e dell’orecchio interno (varie malformazioni cocleovestibolari) (Fig. 5 ). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi (Fig. 6 ) In presenza di questi segni. un’ecografia renale può confermare questa sindrome.. In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell’orecchio esterno, è consigliabile eseguire un’ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1(Abdelhak S., Ruf R.G.). Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale(Vincent C.).
· EYA1 (situato sul cromosoma 8 , [8q13.3]), più di 100 tipi di mutazioni responsabili di BOR, le mutazioni sono divisi in tre gruppi principali (isoforma A, B e C) (presente nel 38% -40,5% dei pazienti)
· Six1 (situato sul cromosoma 14 , [14q23.1]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in 5% dei pazienti),
· SIX5 (situato sul cromosoma 19 , [19q13.32]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in più dell’1% dei pazienti).
Eziologia e la patogenesi
La sindrome BOR viene ereditata come carattere autosomico dominante con elevata penetranza ed espressione variabile. La patogenesi si presume essere una carenza nella differenziazione del primo e secondo arco branchiale. Le anomalie del sistema renale sono il risultato dell’interazione anomala tra la gemma ureterale e il blastema metanefrico. L’orecchio interno (stria vascularis) ed i glomeruli renali condividono un antigene comune, e più di un gruppo di studio ha descritto i cambiamenti istopatologici su esami dell’osso temporale.
|
|
|
|
Fig. 4 : Orecchie a trombetta ed encondromi. |
Un bambino di 13-mesi con sintomi di BOR. L’orecchio sinistro appositamente sviluppato; all’interno l’orecchio destro, il canale uditivo e atresia lobo impropriamente sviluppati. |
|
|
|
|
Fig. 5 : Micrococlea in una sindrome branchio-oto-renale (BOR). |
|
|
|
|
|
Fig. 6 : Residuo branchiale cervicale |
Ragazza con la sindrome di BOR, fistola carotidea bilaterale |
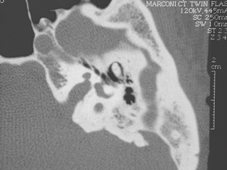
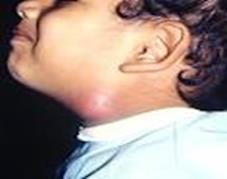
Diagnosi
Una storia familiare positiva per brachiale, otologica, e le malformazioni renali è suggestivo di sindrome di BOR.
Se nessuna storia familiare di malattia è noto, criteri clinici possono essere utilizzate per fare la diagnosi per pazienti con almeno tre criteri principali. In alternativa, la diagnosi può essere fatta per i pazienti che dimostrano due criteri principali e due criteri minori.
Criteri principali
1. Malformazione dei padiglioni auricolari
2. Perdita dell’udito
3. Encondromi preauricolari
4. Anomalie renali
5. Anomalie secondarie dell’arco branchiale
6. Stenosi del canale uditivo esterno
Criteri minori
1. Appendici preauricolari
2. Aplasia del dotto lacrimale
3. Anomalie dell’orecchio medio
4. Anomalie dell’orecchio interno
5. Malformazioni del palato o asimmetria facciale
6. Gozzo eutiroideo
Giudizio fenotipico
Otologico
Le manifestazioni otologiche della BOR sono i sintomi più comuni, con oltre il 90% delle persone in almeno uno dei seguenti:
1. La sordità può presentarsi come trasmissiva, neurosensoriale o mista. La gravità della sordità è variabile, che vanno dalla perdita dell’udito da lieve a profonda. Inoltre, l’entità della perdita dell’udito può essere stabile o progressiva. La perdita dell’udito è ormai consolidata come il tratto singolo più comune tra quelli con la sindrome BOR, stimata in 89% degli individui in uno studio di Fraser et al. Tra quelli con perdita dell’udito, il 50% presentava ipoacusia mista, 30% trasmissiva de il 20% neurosensoriale. E ‘stato inoltre stimato che il 2% dei bambini con sordità profonda hanno BOR.
2. Deformità del padiglione .
3. Bozzi preauricolari.
4. Appendici preauricolari.
5 Stenosi del condotto uditivo esterno:
6. Malformazioni degli ossicini dell’orecchio medio, quali ipoplasia o spostamento
7 Orecchio interno: coclea ipoplasica, canali semicircolari laterali displasici, l’allargamento degli acquedotti vestibolari o cocleari.
Anomalie Arco Branchiale
1. Cisti branchiali leporine.
2. Brancial tratto del seno leporino.
3. Branchiale fistole leporino.
Malformazioni renali
1. Uno spettro di malformazioni renali è possibile, da ipoplasia al agenesia.
2.Cisti caliceale.
3. Uretero-pelvica ostruzione del giunto.
4. Reflusso vescico-ureterale
Altri risultati associati
1. Prolasso della valvola mitrale
2. Palatoschisi
3. Aplasia del dotto lacrimale
4. Paralisi del nervo facciale
5. Retrognazia
Studi dei Geni
Attualmente, vi sono tre mutazioni genetiche conosciuti che provocano sindrome BOR. Se viene effettuata una diagnosi clinica di sindrome di BOR, la conferma dovrebbe essere tentato attraverso l’analisi di sequenza per EYA1. Se una mutazione EYA1 non viene trovato con l’analisi di sequenza, si dovrebbe utilizzare l’analisi duplicazione / cancellazione per EYA1 e analisi di sequenza di SIX 1 e SIX 5.
Mutazione EYA1
· Circa il 40% delle persone con sindrome di BOR avrà una mutazione EYA1.
· EYA1 codifica per un cofattore trascrizione che si esprime nel mesenchima metanefrico durante lo sviluppo dei reni.
SIX1 mutazione
· La mutazione SIX1 è stimata essere trovato 2% dei casi di sindrome BOR.
· Codifica fattori di trascrizione che controllano l’espressione di GDNF e PAX2.
· La maggior parte dei pazienti con questa mutazione non dimostrano malformazione renale.
SIX5 mutazione
· La mutazione SIX5 è presente in circa il 2,5% dei casi di sindrome di BOR.
· Udienza può essere normale in questi pazienti.
Gestione
Valutazione Otologic
· Le prove devono comprendere ABR, audiometria tonale, e la prova di emissione.
· Imaging delle ossa temporali con una TAC è giustificata.
· Una valutazione annuale uditivo dovrebbe essere suggerito.
Valutazione dell’Arco Branchiale
· L’esame fisico seguita dalla tomografia computerizzata (TC), fistologramma, o risonanza magnetica (MRI) se le masse sono palpate .
Valutazione renale
· Ecografia renale
· BUN e creatinina
· I pazienti devono seguire un follow-up annualmente con la nefrologia e urologia.
La consulenza genetica
· È importante che i pazienti affetti da BOR tengano conto conto che c’è una probabilità del 50% di trasmissione per ogni bambino a causa autosomica dominante.
· Test prenatale per BOR è disponibile per quelli con una storia familiare positiva.
Trattamento
Anomalie otologiche
· Rilevare eventuale ipoacusia, utilizzare il trattamento acustico appropriato come indicato dalla gravità della perdita, come la protesi o l’impianto cocleare dell’udito.
· Se l’orecchio medio è intatto, con stenosi del canale uditivo esterno, si può prendere in considerazione una canaloplastica.
· Considerate le procedure di chirurgia plastica per la deformità della Pinna.
Anomalie branchiali
· Asportazione di cisti, fistole, o del seno, con dissezione per tutta la lunghezza del tratto e la tonsillectomia.
Anomalie renali
· Il trattamento dipende dalla gravità delle complicanze renali, ma sia la gestione chirurgica e medica dovrebbe essere utilizzato.
· Il trapianto renale può essere considerato se la malattia renale allo stadio terminale si sviluppa.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Dove posso trovare ulteriori informazioni sulla sindrome branchiootorenale?
Si possono trovare le seguenti risorse sulla sindrome branchiootorenal utili. Questi materiali sono scritti per il pubblico in generale.
• MedlinePlus – Informazioni sanitarie (4 collegamenti)
• Malattie Genetiche e Rare Information Center – Informazioni sulle malattie genetiche e le malattie rare
• Ulteriori NIH Risorse – National Institutes of Health
National Institute on Deafness e altri disturbi della comunicazione (NIDCD)
• Risorse educative – Pagine di informazioni (5 collegamenti)
• Supporto paziente – Per i pazienti e le famiglie (3 collegamenti)
Potreste essere interessati a queste risorse, che sono progettati per gli operatori sanitari e ricercatori.
• Gene Recensioni – Sintesi clinica
• Test genetici Registry – archivio di informazioni test genetico (2 collegamenti)
• PubMed – La letteratura recente
• OMIM – Catalogo Malattia genetica (2 collegamenti)
Quali altri nomi la gente usa per la sindrome branchiootorenale?
• BOR
• Sindrome BOR
• Displasia Branchiootorenale
• Branchio-Otorenale Displasia
• Branchio-Otorenale Syndrome
• Sindrome Branchio-Oto-Renale
• Sindrome di Melnick-Fraser
Per ulteriori informazioni sulla denominazione condizioni genetiche, vedere la Genetics Home Reference Condizione Linee guida di denominazione e di come sono condizioni genetiche e geni denominati? nel manuale.
Cosa succede se ho ancora domande specifiche sulla sindrome branchiootorenal?
Chiedi alla Genetica e Malattie Rare Information Center .
References/Suggested Reading
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M,
Amer I, Falzon A, Choudhury N, Ghufoor K. Branchiootic syndrome-a clinic case report and review of the literature. J Pediatr Surg . 2012;47:1604-1606.
Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet1997;15:157–64.[CrossRef][Medline][Web of Science]
Blanck C, Kohlhase J, Engels S, Burfeind P, Engel W, Bottani A, Patel MS, Kroes HY, Cobben JM. Three novel SALL1 mutations extend the mutational spectrum in Townes-Brocks syndrome: clinical and genetic implications. Am J Hum Genet2000;66:1715–20.[CrossRef][Medline][Web of Science]
Borsani G, DeGrandi A, Ballabio A, Bulfone A, Bernard L, Banfi S, Gattuso C, Mariani M, Dixon M, Donnai D, Metcalfe K, Winter R, Robertson M, Axton R, Brown A, Van Heyningen V, Hanson I. EYA4, a novel vertebrate gene related to Drosophila eyes absent. Hum Mol Genet1999;8:11–23.[Abstract/FREE Full text]
Buck A, Kispert A, Kohlhase J. Embryonic expression of the murine homologue of SALL1, the gene mutated in Townes-Brocks syndrome. Mech Dev2001;104:143–6.[CrossRef][Medline][Web of Science]
Chen A, Francis M, Ni L, Cremers C, Kimberling W, Sato Y, Phelps P, Bellman S, Wagner M, Pembrey M, Smith RJ. Phenotypic manifestations of branchiootorenal syndrome. Am J Med Genet . 1995;58:365-370.
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoot VS, Schwartz CE, Smith RJ. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat . 2004;23:582-9. PubMed citation![]()
Correa-Cerro LS, Kennerknecht I, Just W, Vogel W, Müller D. The gene for branchio-oculo-facial syndrome does not colocalize to the EYA1-4 genes. J Med Genet2000;37:620–3.[FREE Full text]
Engels S, Kohlhase J, McGaughran J. A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet2000;37:458–60.[FREE Full text]
Fraser FC, Sproule JR, Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet . 1980;7:341-349.
Fujimoto A, Lipson M, Lacro RV, Shinno NW, Boelter WD, Jones KL, Wilson MG. New autosomal dominant branchio-oculo-facial syndrome. Am J Med Genet1987;27:943–51.[CrossRef][Medline][Web of Science]
Gene Review: Branchiootorenal Spectrum Disorders This link leads to a site outside Genetics Home Reference. Kochhar A, Fischer SM, Kimberling WJ, et al. Branchio-oto-renal syndrome. Am J Med Genet. 2007;15(143A(14)):1671-8, Review. PubMed citation![]()
Kumar S, Kimberling WJ, Weston MD, Schaefer BG, Berg MA, Marres HA, Cremers CW. Identification of three novel mutations in human EYA1 protein associated with branchio-oto-renal syndrome. Hum Mutat1998;11:443–9.[CrossRef][Medline][Web of Science]
Kumar S, Deffenbacher K, Marres HAM, Cremers CWRJ, Kimberling WJ. Genomewide search and genetic localization of a second gene associated with autosomal dominant branchio-oto-renal J Hum Genet2000;67(suppl 2):401.
Legius E, Fryns JP, Van den Berghe H. Dominant branchial cleft syndrome with characteristics of both branchio-oto-renal and branchio-oculo-facial syndrome. Clin Genet1990;37:347–50.[Medline]
Kumar S, Marres HAM, Cremers CWRJ, Kimberling WJ. Mutation analysis of EYA1 gene associated with
branchio-oto-renal syndrome and refining the BOR2 region on chromosome 1q. Am syndrome. J Med Genet2000;37:303–7.[FREE Full text]
Lin AE, Doherty R, Lea D. Branchio-oculo-facial and branchio-oto-renal syndromes are distinct entities. Clin Genet1992;41:221–3.[Medline]
Lin AE, Gorlin RJ, Lurie IW, Brunner HG, Van der Burgt I, Naumchik IV, Rumyantseva NV, Stengel Rutkowski S, Rosenbaum K, Meinecke P, Müller D. Further delineation of the branchio-oculo-facial syndrome. Am J Med Genet1995;56:42–59.[CrossRef][Medline][Web of Science]
Lin AE, Semina EV, Daack-Hirsch S, Roeder ER, Curry CJ, Rosenbaum K, Weaver DD, Murray JC. Exclusion of the branchio-oto-renal syndrome locus (EYA1) from patients with branchio-oculo-facial syndrome. Am J Med Genet2000;91:387–90.[CrossRef][Medline]
McKusick VA. Mendelian inheritance in man. Catalogs of human genes and genetic disorders. 12th ed. Baltimore: Johns Hopkins University Press, 1998.
Mishima N, Tomarev S. Chicken eyes absent 2 gene: isolation and expression pattern during development. Int J Dev Biol1998;42:1109–15.[Medline][Web of Science]
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes.Proc Natl Acad Sci US A. 2004 May 25;101(21):8090-5.Epub 2004 May 12. PubMed citation![]()
Sahly I, Andermann P, Petit C. The zebrafish eya1 gene and its expression pattern during embryogenesis. Dev Genes Evol1999;209:399–410.[CrossRef][Medline][Web of Science]
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1989. Sanggaard KM, Rendtorff ND, Kjaer KW, Eiberg H, Johnsen T, Gimsing S, Dyrmose J, Nielsen KO, Lage K, Tranebjaerg L. Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses.Eur J Hum Genet.2007 Nov;15(11):1121-31.Epub 2007 Jul 18. PubMed citation![]()
Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet1996;58:1323–37.[Medline][Web of Science]
Vincent C, Kalatzis V, Abdelhak S, Chaib H, Compain S, Helias J, Vaneecloo FM, Petit C. BOR and BO syndromes are allelic defects of EYA1. Eur J Hum Genet1997;5:242–6.[Medline][Web of Science]
Xu PX, Cheng J, Epstein JA, Maas RL. Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci USA1997;94:11974–79.[Abstract/FREE Full text]
Wang Y, Treat K, Schroer RJ O’Brien JE, Stevenson RE, Schwartz CE (giugno 1994). “Localizzazione di (BOR) sindrome Branchio-oto-renale a una regione del cromosoma 3 Mb 8q”. Am. J. Med.. Genet. 51 (2):. 169-75
Sindrome di Stickler/ sindrome di Marshall ,artro-oftalmopatia progressiva ereditaria , displasia di Stickler
distrofia ereditaria artro-oftalmologica
– trasmissione autosomica dominante
– espressività variabile
– sordità neurosensoriale-mista
– palatoschisi
– insufficienza muscolatura faringo-laringea
– miopia
|
Sindrome di Stickler
|
Modalità di trasmissione |
Tipo di deficit uditivo |
Anomalie associate |
|
|
A.D. Gene COL2A1(cr. 12) tipo I, COL11A1 (cr. 1) tipo II, COL11A2 (A.D. e A. R.) |
Ipoacusia neurosensoriale bilaterale piú marcata sulle frequenze acute |
Anomalie ossificazione (epifisi,diafisi), anomalie articolari, ipoplasia facciale, grave miopia (insorgenza primo anno di vita) e frequente distacco di retina, palatoschisi (occasionale). |
CHE COSA È LA SINDROME DI STICKLER??PAG.1
TIPI, PAG.1
STORIA, PERCHÉ SINDROME DI STICKLER?, PAG.2
CAUSE GENETICHE PAG.3
QUALI GENI SONO LEGATI ALLA SINDROME DI STICKLER? PAG.3
EPIDEMIOLOGIA, PAG.4
CAUSE, PAG.4
SINTOMI, PAG.4
LA DIAGNOSI DIFFERENZIALE,PAG10
APPROFONDIMENTI, PAG 17
CHE COSA È LA SINDROME DI STICKLER?
La Sindrome di Stickler (artro-oftalmopatia ereditaria progressiva) è un gruppo di malattie genetiche che interessano il tessuto connettivo , in particolare il collagene . La sindrome di Stickler è un sottotipo di collagenopatia, tipo II e XI Due geni sono stati individuati come responsabili di questa sindrome (COL2A1, COL11 A2), entrambi implicati nella costituzione di certi tipi di collagene. La sindrome può rivelarsi alla nascita con una palatoschisi. micrognatismo, ptosi linguale e soprattutto incompetenza della muscolatura faringo-laringea (turbe della deglutizione ed ostruzione respiratoria) Nelle due varianti cliniche della sindrome, (I e III) è anche presente una forte miopia Durante l’accrescimento diventano evidenti anomalie scheletriche e cartilaginee a carico degli arti. La sordità è neurosensoriale o mista, talvolta complicata da esiti di fiogosi timpaniche croniche.. La sindrome Stickler è caratterizzata da peculiari anomalie facciali, problemi oculari, perdita dell’udito e problemi articolari. E ‘stata studiato per prima e caratterizzato da Gunnar B. Stickler nel 1965.
TIPI
Cambiamenti genetici sono legate ai seguenti tipi di sindrome di Stickler (Annunen S, ed Al 1999; Liberfarb RM ed Al,2003)
Sindrome di Stickler, COL2A1 (75% dei casi Stickler) Fig.1
Sindrome di Stickler, COL11A1
Sindrome di Stickler, COL11A2 (non-oculare)
Sindrome di Stickler, COL9A1 (variante recessiva)
Se ci sono due o tre tipi di sindrome di Stickler è controversa. Ogni tipo è qui presentati per il gene coinvolto. La classificazione di queste condizioni sta cambiando i ricercatori a saperne di più circa le cause genetiche.
|
|
Fig.1 sindrome di Stickler tipo 1 |
I ricercatori hanno descritto diversi tipi di sindrome di Stickler, che si distinguono per la loro causa genetica e loro segni e sintomi caratteristici. In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma IL tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Tipi II e III sono più probabili di tipo I per aver perdita dell’udito. I tipi IV e V sono molto rari e sono stati diagnosticati solo in alcuni individui.
Una condizione simile chiamata sindrome di Marshall è caratterizzata da un aspetto caratteristico del viso, anomalie oculari, perdita dell’udito e artrosi precoce. La Sindrome di Marshall può includere anche bassa statura, che tipicamente non si riscontra nei pazienti con sindrome di Stickler. Se la sindrome di Marshall rappresenta una variante della sindrome di Stickler o una patologia separata è controversa.
Perché sindrome di Stickler?
Stickler sindrome prende il nome dal dottor Gunnar B Stickler. Nel 1960 un ragazzo di dodici anni è stato esaminato presso la Fondazione Mayo nel Minnesota, USA. Il ragazzo aveva ingrandimenti ossee diverse articolazioni ed è stato estremamente miope. Sua madre era totalmente cieco. Dr Stickler ha scoperto che c’erano altri membri della famiglia con sintomi simili, il primo membro della famiglia essendo stati visti dal dottor Charles Mayo nel 1887. Questo ha spinto il dottor Stickler per studiare la famiglia. Con colleghi ha lavorato per definire la condizione, i risultati in corso di pubblicazione nel giugno 1965. Dr Stickler provvisoriamente chiamato la condizione ereditaria PROGRESSIVE ARTRO-OFTALMOPATIA conosciuta in tutto il mondo, come la sindrome di Stickler.
CAUSE GENETICHE
La sindrome deriva da una mutazione di diversi geni del collagene durante lo sviluppo fetale. Si tratta di un sindrome , indipendente dal sesso, autosomica dominante Fig.2, il che significa che un paziente con la sindrome ha una probabilità del 50% di passare la malattia ad ogni figlio. Ci sono tre varianti di Stickler sindrome identificati, ciascuno associato ad un gene di biosintesi del collagene. Un difetto metabolico riguardante l’acido ialuronico e il collagene di tipo 2-d si presuma sia la causa di questa sindrome.
|
|
Fig.2 La sindrome di Stickler è ereditata come modello autosomico dominante |

QUALI GENI SONO LEGATI ALLA SINDROME DI STICKLER?
Le mutazioni nei geni COL11A1, COL11A2 e COL2A1 COL9A1, COL9A2 e geni causano tipi di sindrome di Stickler da I a V, rispettivamente. La sindrome di Marshall, che può essere una variante di Stickler sindrome, risulta da mutazioni nel gene COL11A1. Perché ci siano due o tre tipi di sindrome è controverso. Ciascun tipo è qui presentato secondo il gene in causa. La classificazione di tali condizioni cambierà quando i ricercatori apprenderanno di più circa le cause genetiche. Tali geni sono coinvolti nella produzione del collagene tipo II tipo IX e tipo XI. I collageni sono molecole complesse che provvedono alla struttura e alla resistenza del tessuto connettivo che sostengono le articolazioni del corpo e degli organi. Il collagene di tipo II, tipo IX e XI tipo sono componenti del vitreo, cartilagine e altri tessuti connettivi.. Una mutazione in uno di questi geni interrompe la produzione, l’elaborazione o la formazione del collagene tipo II o XI. Le molecole difettose di collagene o la ridotta quantità di collagene influisce sullo sviluppo di ossa e altri tessuti connettivali, portando al caratteristico quadro della Stickler.
Altri geni, sebbene sconosciuti, possono causare la sindrome perché non tutti i soggetti colpiti hanno mutazioni in uno dei tre geni già identificati. La sindrome di Stickler tipo 1 è dovuta alle mutazioni del gene COL2A1 (12q13.11-q13.2), quella tipo 2 alle mutazioni dei geni COL11A1 (1p21) e COL11A2 (6p21.3). Inoltre, in una famiglia del Marocco, è stata osservata una forma autosomica recessiva associata alle mutazioni del gene COL9A1 (6q12-q14). Le mutazioni che si verificano nel primo gruppo di geni sono a carattere trasmissivo autosomico dominante mentre quelle a carico del secondo gruppo sono a carattere autosomico recessivo e, in entrambi i casi, le conseguenze fenotipiche che comportano sono molte.
EPIDEMIOLOGIA
Complessivamente la prevalenza stimata di sindrome Stickler è circa 1 su 10.000. Colpisce 1 neonato su 7.500-9.000. Alcuni medici credono che ben 3 in 10.000 persone sono colpite, ma ulteriori ricerche sono necessarie per confermare questo. Come una malattia ereditaria, la sindrome di Stickler è normalmente passata di padre in figlio. C’è una probabilità del 50% dei bambini essere colpiti in questo modo anche se ci sono alcuni casi registrati dove si è verificato per la prima volta in un bambino.
CAUSE
La sindrome è ereditaria con schema autosomico dominante. Si suppone che insorga da una mutazione di diversi geni del collagene durante lo sviluppo fetale. È indipendente dal sesso, cioè i soggetti affetti dalla sindrome hanno la probabilità del 50% di passarla a ciascun figlio.
SINTOMI
Gli individui con sindrome di Stickler hanno una serie di segni e sintomi. Alcune non hanno segni e sintomi; altri hanno alcune o tutte le funzioni descritte di seguito. Inoltre, ogni caratteristica di questa sindrome può variare da una forma lieve ad una grave.
Fu studiata e catalogata per la prima volta dal dr. G.B. Stickler nel 1965. È caratterizzata dal
1) tipico aspetto facciale appiattito, Fig. 3
2) patologie oculari,
3) perdita dell’udito,
4)disturbi alle articolazioni.
1)tipico aspetto facciale appiattito: Fig. 3A-B-C-D-E questo è causato da ossa sottosviluppate del centro del viso, compresi gli zigomi e il ponte del naso. Un gruppo particolare di caratteristiche fisiche, chiamata sequenza di Pierre Robin, è comune anche in persone con sindrome di Stickler. La sequenza Robin include un’apertura nel tetto della bocca (palatoschisi), una lingua piccola , che è posto più indietro rispetto al normale (glossoptosi), e una piccola mandibola (micrognazia). Questa combinazione di caratteristiche può portare a problemi di alimentazione e difficoltà di respirazione.. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante.
|
|
|
||||
|
Fig. 3A-B-C-D-E |
|
||||
|
|
||||
|
|
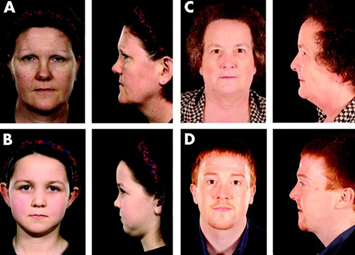


2)patologie oculari,
Miopia
Alto rischio di distacchi di retina, che possono influenzare entrambi gli occhi.
Cataratta
Glaucoma
La sindrome di Stickler è una vitreo-retinopatia. Anomalie di formazione vitreo e dell’architettura gel sono patognomonico della sindrome di Stickler e, a nostro avviso un prerequisito per la diagnosi. 6 Due distinti fenotipi possono essere riconosciute. 7-9La maggior parte dei pazienti hanno una caratteristica anomalia congenita del vitreo (il tipo 1 fenotipo, Fig 4 A ) e questo correla con difetti di di procollagene di tipo II.78 Un gel vitreale apparentemente rudimentale occupa lo spazio immediato retrolentale ed è delimitato da una membrana distinta piegata. In una minoranza di pedigree c’è un fenotipo diverso con fasci radi e irregolarmente ispessite di fibre in tutta la cavità vitrea (tipo 2 fenotipo, Fig. 4B).
|
|
|
|
Fig4 A : fenotipo tipo 1, |
Fig. 4B :fenotipo, tipo 2 |

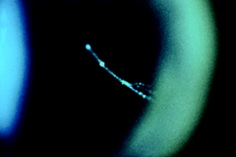
In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma il tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Molte persone con sindrome di Stickler sono molto miopi (descritto ed aventi elevata miopia ) a causa della forma dell’occhio. Anomalie dello sviluppo dell’angolo di drenaggio camera anteriore predispongono i pazienti a glaucoma, 10Fig. 7 (b), ma la complicanza più grave oftalmica riferisce l’elevato rischio di distacco di retina, di solito come conseguenza di gigante formazione di rottura retinica Può Anche presente come complicazione la cataratta associata alla sindrome di Stickler. La sostanza gelatinosa all’interno dell’occhio (il vitreo ) ha un aspetto distintivo dei tipi di sindrome di Stickler associati ai geni COL2A1 e COL11A1. Come risultato si consiglia appuntamenti fissi ad un oculista specialista. Il tipo di sindrome di Stickler associata al gene COL11A2 non influenza l’occhio. [2]
|
|
|
Tutti i pazienti hanno mostrato il fenotipo vitreo “perline” (Fig. 4 C-D-E) ed hanno confermato le mutazioni nella catena α1 di tipo XI collagene ( COL11A1 ) |
|
|
|
Fig. 5 A-B-C Caratteristica cataratta corticale |
|
|
|
Fig. 6 (A) lieve opacità della capsula posteriore del cristallino dell’occhio destro osservata inizialmente all’età di 7 anni. (B) Aspetto del fondo dimostrare la retina alterazioni della pigmentazione della periferia e alterazioni degenerative della retina all’interno del polo posteriore. (C) opacizzazione marcata e la fusione delle capsule lente nell’occhio di destra osservato a 9 anni di età. (D) Lievi opacità della capsula si osservano ancora due anni più tardi, a 11 anni di età. L’errore di rifrazione in questa fase è +1.25 e l’acuità visiva per una distanza di 6/12 (20/40). |


3)perdita dell’udito. La sordità di percezione, trasmissione o mista è incostante, e spesso mascherata o aggravata dai problemi di otite cronica che sono associati all’incompetenza faringea. Essa è spesso evolutiva. Sono descritti tre tipi di sindrome di Stickler: nei tipi 1 e 3, agli altri elementi della sindrome è associata una miopia molto forte con rischio di degenerazione vitro-retinica. L’esame oftalmologico di ogni bambino sordo e di ogni lattante con incompetenza faringolaringea deve permettere d’individuare questa sindrome e correggere precocemente la miopia. La maggior parte delle persone con sindrome di Stickler hanno anomalie scheletriche che colpiscono le articolazioni. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
4)disturbi alle Ossa e Articolazioni
Caratterizzata dall’associazione tra segni oculari, correlati alle forme più o meno complete della sequenza di Pierre Robin (si veda questo termine), alterazioni scheletriche e sordità neurosensoriale (10% dei casi) .Questi segni e sintomi variano ampiamente tra gli individui affetti. Questa sindrome è dovuta a una alterazione delle catene ![]() di alcuni collageni. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare di tipo artrosico o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
di alcuni collageni. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare di tipo artrosico o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
I ricercatori hanno descritto diversi tipi di sindrome di Stickler, che si distinguono per la loro causa genetica e loro segni e sintomi caratteristici. In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma il tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Nei tipi II e III la perdita dell’udito è più probabili rispetto al tipo I . I tipi di IV e V sono molto rari e sono stati diagnosticati solo in alcuni individui.
Una simile condizione chiamata sindrome di Marshall è caratterizzata da un aspetto caratteristico del viso, anomalie oculari, perdita dell’udito e artrosi precoce. Sindrome di Marshall può includere anche bassa statura, che tipicamente non si riscontra nei pazienti con sindrome di Stickler. E’ controverso se la sindrome di Marshall rappresenti una variante della sindrome di Stickler o piuttosto n’entità a se.
|
|
|
|
|
Fig. 7 (A) Miopia. |
Fig. 7 (b) Foto che mostra vasi congiuntivali dilatati a bordo corneale (flush ciliare, filo circumcorneale) e cornea nebbiosa caratteristica di glaucoma acuto ad angolo chiuso |
|
|
|
|
|

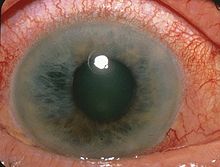
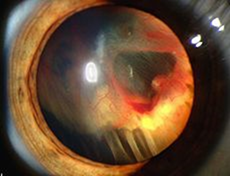
Le persone con sindrome di Stickler accusano una serie di segni e sintomi, dalla paucità alla grave espressione fenotipica.
Una tipica caratteristica della sindrome è l’aspetto della faccia, come appiattito. Ciò è causato dalle ossa poco sviluppate nel centro della faccia, comprese le mascelle e il setto del naso.
Molti pazienti con la sindrome di Stickler soffrono di la cataratta giovanile, miopia grave Fig. 7 (A) , a causa della forma dell’occhio. Gli individui con gli occhi colpiti dalla patologia sono soggetti a pressione oculare aumentata (glaucoma) e a degenerazione vitreo-retinica o corio-retinica, a distacco della retina e l’uveite cronica.. La sostanza gelatinosa dentro l’occhio ha un aspetto caratteristico nella sindrome di tipo COL2A1 e COL11A1. Quella di tipo COL11A2 non colpisce l’occhio.
I soggetti con questa patologia hanno disturbi che colpiscono anche altri organi. Artrite, anormalità dell’estremità delle ossa lunghe, anomalia delle vertebre, curvatura della spina dorsale, gibbosità, dolore all’articolazioni,
ginocchio valgo sono tutti problemi che possono occorrere alle ossa e alle articolazioni. L’artrite spesso compare a giovane età e peggiora con la crescita).Le articolazioni dei bambini colpiti e dei giovani possono essere molto flessibili (ipermobili).. Difficoltà nell’apprendimento possono anche avvenire per la menomazione all’udito e alla vista.Tipico di soggetti con Stickler può includere guance piatte, setto nasale appiattito e mascella piccola, anomalie del palato.
Un altro segno della sindrome è la diminuzione dell’udito da lieve a grave che, per alcuni, può essere progressiva. La perdita di udito è stata riscontrata nel 62,9 per cento dei casi, in una forma lieve o moderata. Il deficit uditivo era principalmente neurosensoriale ovvero a carico della coclea o del nervo acustico (67,8 per cento).
Il deficit trasmissivo, danno a carico dell’orecchio esterno o medio, è risultato essere meno frequente (14,1 pe cento) cosi come quello misto (18,1 per cento). Le forme di ipoacusia trasmissiva e mista sono state riscontrate principalmente nei giovani pazienti o nei pazienti con un difetto al palato.
Complessivamente le mutazioni a carico di COL11A1 (82,5 per cento) e COL11A2 ( 94,1 per cento) risultano più frequentemente associate con la menomazione uditiva, rispetto alle mutazioni di COL2A1 ( 52,2 per cento Considerando questi dati si può sostenere che la menomazione uditiva nei pazienti affetti da sindrome di Stickler è comune e che, nonostante la forma di ipoacusia che predomini sia quella neurosensoriale, anche l’ipoacusia trasmissiva e mista possono verificarsi.
Le mutazioni dei diversi geni codificanti per il collagene sono associate a diversi tassi di prevalenza di ipoacusia ma esistono ancora un gran numero di variazioni fenotipiche che non sono state associate a specifiche caratteristiche genotipiche.
Vista la portata del problema il consiglio è di eseguire un regolare follow-up dell’udito nei pazienti affetti da sindrome di Stickler, valutazioni audiologiche ogni sei mesi fino all’età cinque anni, poi annualmente,
La diagnosi differenziale
Diversi disturbi simili alla sindrome di Stickler sono stati descritti e il loro status di entità distinte rimane controversa. Dati genetici molecolari stanno cominciando a informare questo dibattito ma resta incertezza. Questo sarà risolto solo quando più dati di genotipo diventano disponibili in combinazione con la descrizione dettagliata dei oculare associata e fenotipo non-oculare.
SINDROME DI WAGNER
Wagner29ha registrato una grande famiglia svizzera con un disordine autosomico dominante dell’occhio assomiglia sindrome di Stickler, ma senza il distacco di retina. Molte delle famiglie successivamente segnalato come sindrome di Wagner hanno avuto caratteristiche sistemiche in comune con la sindrome di Stickler e la distinzione tra le due condizioni è diventata sfocata. Infatti, alcuni autori hanno suggerito che essi sono la stessa malattia. 30La prova che le famiglie che mostrano solo le manifestazioni oculari della sindrome di Wagner hanno una condizione distinta dalla sindrome di Stickler è venuto dalla constatazione di linkage to 5q13-q14 nella famiglia di origine Wagner 31e esclusione di linkage to COL2A1 in un’altra famiglia.32Alla luce di questi risultati, il termine “sindrome di Wagner-Stickler” dovrebbe essere abbandonato. Korkko et al33ha riferito di un paziente con la sindrome di Wagner derivante da una sostituzione di ingombranti aspartato aminoacido glicina per nell’esone 10 di COL2A1 e postulato un possibile legame tra il tipo di mutazione e fenotipi Stickler o Wagner. Tuttavia, frequente distacco della retina e, in misura minore cataratta sono stati attribuiti alla sindrome di Wagner, mentre nel report originale 29nessun paziente ha subito un distacco della retina, “cataracta Complicata” è stata quasi universale, e la miopia era bassa in tutti i casi. Si potrebbe sostenere che nella famiglia riportata da Korkko et al 33fenotipo assomiglia più da vicino la sindrome di Stickler sindrome di Wagner.
EROSIVA VITREORETINICA
Brown et al 34 hanno descritto un disordine autosomico occhio dominante hanno chiamato vitreoretinica erosiva con un fenotipo simile sindrome di Wagner e privo di eventuali anomalie sistemiche. Questa condizione è stato mappato a 5q13-q14 suggerendo che potrebbe essere una variante allelica della sindrome di Wagner. 31
SINDROME DI MARSHALL
Marshall 35ha registrato una grande famiglia che mostra ereditarietà autosomica dominante della cataratta, miopia, vitreo anormale, ipoplasia mediofacciale e sordità congenita. Marshall ha pensato il fenotipo potrebbe rappresentare una forma incompleta di ereditaria anidrotica displasia ectodermica, ma ha riconosciuto che il pelo era normale e la prova di ipodonzia e ipoidrosi “non era fortemente convincente”. Shanske et al 36ha notato dalle foto pubblicate che uno dei pazienti di Marshall era colpito da ipertelorismo con ipertelorismo forse lieve in altri. C’è stato molto dibattito se la sindrome di Marshall è una entità distinta 37 e, in caso affermativo, se displasia ectodermica è una caratteristica della condizione. 36Ayme e Preus 38 effettuate cluster analysis sui rapporti pubblicati di Marshall e Stickler sindrome pazienti e ha concluso che erano diversi. Resta da vedere se questo problema può essere risolto con l’analisi genetica molecolare. Griffith et al 39hanno riportato una mutazione COL11A1 in una famiglia ha detto di avere la sindrome di Marshall. Shanske et al40hanno suggerito la famiglia aveva la sindrome di Stickler, ma gli autori hanno risposto che, come riportato da Marshall famiglia di origine, i loro pazienti avevano cataratta congenita e giovanile, vitreo fluido, perdita di udito, e simili aspetto cranio-facciale e la radiologia. 41,in quanto questi sono tutte le caratteristiche della sindrome di Stickler riconosciuto e non vi è alcuna informazione sul fenotipo vitreo, il problema rimane irrisolto.
SINDROME DI WEISSENBACHER-ZWEYMÜLLER E OSMED
Weissenbacher e Zweymuller 42hanno descritto un neonato con la sequenza di Pierre-Robin, naso camuso, prossimale accorciamento dell’arto, femori a forma di campana muta e omeri, e schisi vertebrali coronali. I genitori erano sani e non collegati. Giedion et al 43hanno seguito lo stesso paziente a 18 anni di età. Sordità neurosensoriale aveva sviluppato all’età di 5. By adulta accorciamento della vita arto era del tutto risolto e l’altezza e l’aspetto erano sostanzialmente normale. Non c’era alcuna anomalia occhio. Epifisi ingrossati erano una caratteristica radiologica di primo piano in adolescenza. Giedion et al 43hanno riferito di altri tre pazienti con lo stesso fenotipo e ha coniato il nome OtoSpondiloMegaEpifiseale Displasia (OSMED). Essi hanno concluso che la sindrome Weissenbacher-Zweymuller (WZS) e OSMED erano la stessa sindrome. Pihlajamaa et al 44,successivamente è emerso che il paziente WZS originale era eterozigoti per una mutazione in COL11A2. Sono state descritte altre famiglie mostrano autosomica dominante di un simile non-oculare Stickler sindrome fenotipo come risultato di mutazioni COL11A2. 45-47 Van Steensel et al 48riportano tre fratelli dei genitori consanguinei che erano omozigoti per una mutazione COL11A2 e aveva il OSMED fenotipo. Vikkula et al 46hanno riportato diversi membri di una famiglia consanguinea che erano omozigoti per una mutazione in COL11A2. Avevano una grave sordità neurosensoriale congenita, ipoplasia medio-facciale, a breve, naso all’insù, gli occhi prominenti, creste sopraorbitali prominenti, e insorgenza precoce adulta di grave artrosi delle anche, ginocchia, spalle e gomiti. Altezza adulta è stato leggermente ridotto con l’aumento della lordosi lombare. Le articolazioni interfalangee erano prominenti con brevi quinto metacarpo. Come in tutte le famiglie segnalate con mutazioni COL11A2, esame oftalmologico era normale.
ALTRI DISTURBI
Altre malattie con alcune caratteristiche in comune con la sindrome di Stickler includono displasia congenita spondiloepifiseale, 49 displasia di Kniest, 49-51 e la sindrome di Marfan. 52
References
1. ↵
Stickler GB, Belau PG, Farrell FJ, et al.(1965) Hereditary progressive arthro-ophthalmopathy. Mayo Clinic Proc 40:433–455.[Medline][Web of Science]Google Scholar
2. ↵
1. Stickler GB, Pugh DG(1967) Hereditary progressive arthro-ophthalmopathy. II. Additional observations on vertebral abnormalities, a hearing defect, and a report of a similar case. Mayo Clinic Proc 42:495–500.[Web of Science]Google Scholar
3. ↵
1. Temple IK(1989) Stickler’s syndrome. J Med Genet 26:119–126.[FREE Full text]
4. ↵
1. Scott JD(1989) Duke-Elder lecture. Prevention and perspective in retinal detachment. Eye 3:491–515.Google Scholar
5. ↵
1. Seery CM, Pruett RC, Liberfarb RM, Cohen BZ(1990) Distinctive cataract in the Stickler syndrome. Am J Ophthalmol 110:143–148.[Medline][Web of Science]Google Scholar
6. ↵
1. Maumenee IH(1979) Vitreoretinal degeneration as a sign of generalized connective tissue diseases. Am J Ophthalmol 88:432–449.[Medline][Web of Science]Google Scholar
7. ↵
1. Snead MP, Payne SJ, Barton DE, et al.(1994) Stickler syndrome: correlation between vitreoretinal phenotypes and linkage to COL 2A1. Eye 8:609–614.Google Scholar
8. ↵
1. Snead MP, Yates JR, Pope FM, Temple IK,Scott JD.(1996) Masked confirmation of linkage between type 1 congenital vitreous anomaly and COL 2A1 in Stickler syndrome. Graefes Arch Clin Exp Ophthalmol 234:720–721.[CrossRef][Medline][Web of Science]Google Scholar
9. ↵
1. Snead MP(1996) Hereditary vitreopathy. Eye 10:653–663.Google Scholar
10. ↵
1. Nielson CE.(1981) Stickler’s syndrome. Acta Ophthalmol 59:286–295.Google Scholar
11. ↵
1. Cho H,Yamada Y, Yoo TJ.(1991) Ultrastructural changes of cochlea in mice with hereditary chondrodysplasia (cho/cho). Ann N Y Acad Sci 630:259–261.[Medline][Web of Science]Google Scholar
12. ↵
1. Lucarini JW, Liberfarb RM,Eavey RD(1987) Otolaryngological manifestations of the Stickler syndrome. Int J Pediatr Otorhinolaryngol 14:215–222.[CrossRef][Medline][Web of Science]Google Scholar
13. ↵
1. Beighton P(1993) McKusick’s heritable disorders of connective tissue. (Mosby, St Louis), 5th ed..Google Scholar
14. ↵
1. Rai A, Wordsworth P, Coppock JS, Zaphiropoulos GC, Struthers GR.(1994) Hereditary arthro-ophthalmopathy (Stickler syndrome): a diagnosis to consider in familial premature osteoarthritis. Br J Rheumatol 33:1175–1180.[Abstract/FREE Full text]
15. ↵
1. Beals RK.(1977) Hereditary arthro-ophthalmopathy (the Stickler syndrome). Report of a kindred with protrusio acetabuli. Clin Orthop 125:32–35.Google Scholar
16. ↵
1. Liberfarb RM, Goldblatt A.(1986) Prevalence of mitral-valve prolapse in the Stickler syndrome. Am J Med Genet 24:387–392.[CrossRef][Medline][Web of Science]Google Scholar
17. ↵
1. Prockop DJ, Kivirikko KI, Tuderman L, Guzman NA.(1979) The biosynthesis of collagen and its disorders (second of two parts). N Engl J Med 301:77–85.[Medline][Web of Science]Google Scholar
18. ↵
1. Prockop DJ, Kivirikko KI, Tuderman L,Guzman NA.(1979) The biosynthesis of collagen and its disorders (first of two parts). N Engl J Med 301:13–23.[CrossRef][Medline][Web of Science]Google Scholar
19. ↵
1. Francomano CA.(1995) Key role for a minor collagen. Nat Genet 9:6–8.[CrossRef][Medline][Web of Science]Google Scholar
20. ↵
1. Pope FM.(1998) Molecular abnormalities of collagen and connective tissue. Maddison PJ, Isenberg DA, Woo P, Glass DN, editors. Oxford textbook of rheumatology. (Oxford University Press, Oxford), pp 353–404.Google Scholar
21. ↵
1. Prockop DJ, Kivirikko KI.(1995) Collagens: molecular biology, diseases and potentials for therapy. Annu Rev Biochem 64:403–434.[CrossRef][Medline][Web of Science]Google Scholar
22. ↵
1. Upholt WB, Strom CM, Sandell LJ.(1985) Structure of the type II collagen gene. Ann N Y Acad Sci 460:130–140.[CrossRef][Medline][Web of Science]Google Scholar
23. ↵
1. Ballo R, Beighton PH, Ramesar RS.(1998) Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am J Med Genet 80:6–11.[CrossRef][Medline][Web of Science]Google Scholar
24. ↵
1. Horton WA.(1996) Progress in human chondrodysplasias: molecular genetics. Ann N Y Acad Sci 785:150–159.[Medline]Google Scholar
25. ↵
1. Richards AJ, Yates JR, Williams R, et al.(1996) A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet 5:1339–1343.[Abstract/FREE Full text]
26. ↵
1. Martin S, Richards AJ,Yates JRW, Pope FM, Scott JD, Snead MP.(1998) Stickler syndrome types 1 and 2: confirmation of genetic heterogeneity and evidence for another locus. Presentation XXI Meeting Club Jules Gonin, p A34.Google Scholar
27. ↵
1. Mayne R, Brewton RG, Mayne PM, Baker JR.(1993) Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J Biol Chem 268:9381–9386.[Abstract/FREE Full text]
28. ↵
1. Wilkin DJ, Mortier GR, Johnson CL, et al.(1998) Correlation of linkage data with phenotype in eight families with Stickler syndrome. Am J Med Genet 80:121–127.[CrossRef][Medline][Web of Science]Google Scholar
29. ↵
1. Wagner H.(1938) Ein bisher unbekanntes Erbleiden des Auges (Degeneratio hyaloideo-retinalis hereditaria), beobachtet im Kanton Zurich. Klin Monatsbl Augenheilkd 100:840–857.Google Scholar
30. ↵
1. Liberfarb RM, Hirose T, Holmes LB.(1981) The Wagner-Stickler syndrome: a study of 22 families. J Pediatr 99:394–399. [CrossRef][Medline][Web of Science]Google Scholar
31. ↵
1. Brown DM, Graemiger RA, Hergersberg M, et al.(1995) Genetic linkage of Wagner disease and erosive vitreoretinopathy to chromosome 5q13-14. Arch Ophthalmol 113:671–675.[CrossRef][Medline][Web of Science]Google Scholar
32. ↵
1. Fryer AE, Upadhyaya M ,Littler M, et al.(1990) Exclusion of COL2A1 as a candidate gene in a family with Wagner-Stickler syndrome. J Med Genet 27:91–93.[Abstract/FREE Full text]
33. ↵
1. Korkko J, Ritvaniemi P, Haataja L, et al.(1993) Mutation in type II procollagen (COL2A1) that substitutes aspartate for glycine alpha 1-67 and that causes cataracts and retinal detachment: evidence for molecular heterogeneity in the Wagner syndrome and the Stickler syndrome (arthro-ophthalmopathy). Am J Hum Genet 53:55–61.[Medline]Google Scholar
34. ↵
1. Brown DM, Kimura AE, Weingeist TA, Stone EM.(1994) Erosive vitreoretinopathy. A new clinical entity. Ophthalmology 101:694–704.[Medline][Web of Science]Google Scholar
35. ↵
1. Marshall D.(1958) Ectodermal dysplasia. Report of a kindred with ocular deformities and hearing defect. Am J Ophthalmol 45:143–156.[Medline][Web of Science]Google Scholar
36. ↵
1. Shanske AL, Bogdanow A, Shprintzen RJ, Marion RW.(1997) The Marshall syndrome: report of a new family and review of the literature. Am J Med Genet 70:52–57.[CrossRef][Medline]Google Scholar
37. ↵
1. Cohen MM, Jr.(1974) The demise of the Marshall syndrome. J Pediatr 85:878.[Medline]Google Scholar
38. ↵
1. Ayme S, Preus M.(1984) The Marshall and Stickler syndromes: objective rejection of lumping. J Med Genet 21:34–38.[Abstract/FREE Full text]
39. ↵
1. Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH,Warman ML.(1998) Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am J Hum Genet 62:816–823.[CrossRef][Medline][Web of Science]Google Scholar
40. ↵
1. Shanske A, Bogdanow A, Shprintzen RJ, Marion RW.(1998) Marshall syndrome and a defect at the COL11A1 locus. Am J Hum Genet 63:1558–1561.[CrossRef][Medline][Web of Science]Google Scholar
41. ↵
1. Warman ML, Tiller GE, Griffith AJ.(1998) Reply to Shanske et al. Am J Hum Genet 63:1559–1561.[CrossRef][Medline][Web of Science]Google Scholar
42. ↵
1. Weissenbacher G, Zweymuller E.(1964) Coincidental occurrence of Pierre Robin and foetal chondrodysplasia. Monatsschr Kinderheilkd 112:315–317.[Medline]Google Scholar
43. ↵
1. Giedion A, Brandner M, Lecannellier J, et al.(1982) Oto-spondylo-megaepiphyseal dysplasia (OSMED). Helv Paediatr Acta 37:361–380.[Medline]Google Scholar
44. ↵
1. Pihlajamaa T, Prockop DJ, Faber J,et al.(1998) Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am J Med Genet 80:115–120.[CrossRef][Medline][Web of Science]Google Scholar
45. ↵
1. Brunner HG, van Beersum SE, Warman ML, Olsen BR, Ropers HH, Mariman EC(1994) A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet 3:1561–1564.[Abstract/FREE Full text]
46. ↵
1. Vikkula M, Mariman EC, Lui VC, et al.(1995) Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 80:431–437.[CrossRef][Medline][Web of Science]Google Scholar
47. ↵
1. Sirko-Osadsa DA, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH.(1998) Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J Pediatr 132:368–371.[CrossRef][Medline][Web of Science]Google Scholar
48. ↵
1. van Steensel MA, Buma P, de Waal Malefijt MC, van den Hoogen FH, Brunner HG.(1997) Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. Am J Med Genet 70:315–323.[CrossRef][Medline][Web of Science]Google Scholar
49. ↵
1. Spranger J, Winterpacht A, Zabel B.(1994) The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr 153:56–65.[Medline][Web of Science]Google Scholar
50. ↵
1. Kniest W.(1952) Zur abgrenzung der dysostosis enchondrallis von der chondrodystrophie. Z Kinderheilkd 70:633–640.[CrossRef][Medline]Google Scholar
51. ↵
1. Maumenee IH, Traboulsi EI.(1985) The ocular findings in Kniest dysplasia. Am J Ophthalmol 100:155–160.[Medline][Web of Science]Google Scholar
52. ↵
1. Gray JR, Davies SJ.(1996) Marfan syndrome. J Med Genet 33:403–408.[FREE Full text]
53. ↵
1. Brown DM, Vandenburgh K, Kimura AE, Weingeist TA, Sheffield VC, Stone EM.(1995) Novel frameshift mutations in the procollagen 2 gene (COL2A1) associated with Stickler syndrome (hereditary arthro-ophthalmopathy). Hum Mol Genet 4:141–142.[FREE Full text]
54. ↵
1. Ahmad NN, McDonald-McGinn DM, Zackai EH,et al.(1993) A second mutation in the type II procollagen gene (COL2AI) causing Stickler syndrome (arthro-ophthalmopathy) is also a premature termination codon. Am J Hum Genet 52:39–45.[Medline][Web of Science]Google Scholar
55. ↵
1. Williams CJ, Ganguly A, Considine E, et al.(1996) A-2→G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred. Am J Med Genet 63:461–467.[CrossRef][Medline][Web of Science]Google Scholar
56. ↵
1. Ahmad NN, Ala-Kokko L, Knowlton RG, et al.(1991) Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA 88:6624–6627.[Abstract/FREE Full text]
57. ↵
1. Brown DM, Nichols BE, Weingeist TA, Sheffield VC, Kimura AE,Stone EM.(1992) Procollagen II gene mutation in Stickler syndrome. Arch Ophthalmol 110:1589–1593.[CrossRef][Medline][Web of Science]Google Scholar
58. ↵
1. Ritvaniemi P, Hyland J, Ignatius J, Kivirikko KI, Prockop DJ, Ala-Kokko L.(1993) A fourth example suggests that premature termination codons in the COL2A1 gene are a common cause of the Stickler syndrome: analysis of the COL2A1 gene by denaturing gradient gel electrophoresis. Genomics 17:218–221.[CrossRef][Medline][Web of Science]Google Scholar
59. ↵
1. Ahmad NN, Dimascio J, Knowlton RG,Tasman WS.(1995) Stickler syndrome. A mutation in the nonhelical 3′ end of type II procollagen gene. Arch Ophthalmol 113:1454–1457.[CrossRef][Medline][Web of Science]Google Scholar
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Didascalia : |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Categorie DFN
Categorie DFN
Le sordità
genetiche nella maggioranza dei casi sono malattie “mono-geniche”:
l’alterazione di un solo gene è responsabile di ciascuna forma di sordità, e la
sordità è per lo più causata da un disordine dell’orecchio interno. Le diverse
forme di sordità non sindromica sono state caratterizzate per mezzo di un
analisi di legame genetico (“linkage”) eseguita su famiglie numerose che permettono
di definire un “locus” su uno dei cromosomi, cioè una regione genomica
contenente il gene responsabile della sordità. Spesso il gene viene isolato in
un secondo tempo, ed in qualche caso un singolo locus può contenere due geni
della sordità.
Una convenzione internazionale ha stabilito di denominare i loci della sordità
non sindromica con le sigle DFNA (deafness, autosomica dominante loci 1-41),
DFNB (deafness, autosomica recessiva loci 1-33), e DFN (deafness, X-linked loci
1-8) Tali sigle sono seguite da un numero, che definisce l’ordine di tempo nel
quale ogni forma è stata definita. Ad oggi esistono 60 forme (24 con geni
donati), 80 forme DFNB (39 con geni donati), e 5 forme DFN, Oltre a queste,
nella classificazione sono state inserite 2 forme DFNM, (M modificante)
caratterizzate dalla presenza di un gene “modificante”, cioè in grado di
cambiare l’espressività di una determinata mutazione genetica. Le relazioni fra
geni, fenotipo, e modalità di trasmissione del carattere sono complesse secondo
il tipo di mutazione: così uno stesso gene può determinare una DFNA e/o una
DFNB, e/o una sordità sindromica.
Nel
50% dei casi di sordità asindromica autosomica recessiva la causa è
rappresentata da mutazioni presenti nella regione genomica (locus) DFNB1
presente sul braccio lungo del cromosoma 13. Il locus DFNB1 contiene il gene
GJB2 (Gap Junction-2; OMIM 121011) che codifica per la proteina Connessina 26,
ed il gene GJB6 (OMIM 604418) che codifica per la proteina Connessina 30. Ad
oggi sono state individuate circa 100 mutazioni a carico del gene GJB2
(http://davinci.crg.es/deafness) e 2 delezioni che coinvolgono il gene GJB6
(D13S1830 e D13S1854) e che sono la causa della sordità non sindromica legata
al locus DFNB1.
Fino ad oggi si sono identificati circa 150 “loci” cromosomici associati a
forme genetiche di sordità. Si stima che questo numero rappresenti solamente la
metà delle alterazioni genetiche che potrebbero causare una sordità. Esistono
database che aggiornano puntualmente la classificazione delle sordità genetiche
(per es.: hffp://hereditaryhearinglossorg).
La
maggior parte delle forme di sordità non sindromica sono associati con perdita
di udito permanente causata da danni alle strutture nell’orecchio interno.
L’orecchio interno è costituito da tre parti: una struttura a forma di
chiocciola chiamata coclea che aiuta suono processo, nervi che inviano informazioni
dalla coclea al cervello, e strutture coinvolte con equilibrio. La perdita
dell’udito causata da cambiamenti dell’orecchio interno è chiamato sordità
neurosensoriale. La perdita dell’udito che deriva da variazioni nel
dell’orecchio medio è chiamato ipoacusia trasmissiva. L’orecchio medio contiene
tre piccole ossa che aiutano il suono trasferimento dal timpano all’orecchio
interno. Alcune forme di sordità non sindromiche comportano cambiamenti sia
l’orecchio interno e l’orecchio medio; questa combinazione viene chiamata
ipoacusia mista.
La
gravità della perdita dell’udito varia e può cambiare nel tempo. Può
interessare un orecchio (unilaterale) o entrambe le orecchie (bilaterali).
Gradi di gamma perdita dell’udito da lieve (difficoltà a comprendere il
linguaggio morbido) a profonda (incapacità di sentire anche rumori molto
forti). La perdita può essere stabile, o può progredire come una persona
invecchia. Particolari tipi di sordità sindromica spesso mostrano modelli
distintivi di perdita dell’udito. Ad esempio, la perdita può essere più
pronunciato ad alta, media o bassa tonalità.
Epidemiologia
Circa 1 su
1000 bambini negli Stati Uniti nasce con sordità profonda. In 9 anni,
circa 3 su 1.000 bambini hanno perdita che colpisce le attività della vita
quotidiana dell’udito. Più della metà di questi casi sono causati da
fattori genetici. La maggior parte dei casi di sordità genetica (70% al
80%) sono non sindromiche; restanti casi sono causati da sindromi
genetiche specifiche. Negli adulti, la possibilità di sviluppare perdita
dell’udito aumenta con l’età; perdita dell’udito colpisce la metà di tutte
le persone di età superiore ai 80 anni.
Geni
legati alla sordità non sindromica
Mutazioni
in the ACTG1 , CDH23 , CLDN14 , COCH , COL11A2 , DFNA5 , ESPN , EYA4 , GJB2 , GJB6 , KCNQ4 , MYO15A , MYO6 ,MYO7A , OTOF , PCDH15 ,
POU3F4 , SLC26A4 , STRC , TECTA , TMC1 , TMIE , TMPRSS3 , USH1C , e WFS1
geni causano sordità non
sindromica, con prove più deboli attualmente implicando genes
CCDC50 , DIAPH1 , DSPP , ESRRB , GJB3 , GRHL2 , GRXCR1 , HGF ,LHFPL5 , LOXHD1 , LRTOMT , MARVELD2 , MIR96 , MYH14 , MYH9 ,
MYO1A , MYO3A , OTOA , PJVK , POU4F3 , PRPS1 , PTPRQ ,RDX , SERPINB6 , SIX1 , SLC17A8 , TPRN , TRIOBP , e whrn.
Le cause di
sordità non sindromiche possono essere complesse. I ricercatori hanno
identificato più di 30 geni che, se mutati, possono provocare la sordità non
sindromica; Tuttavia, alcuni di questi geni non sono state pienamente
caratterizzato. Molti geni legati alla sordità sono coinvolti nello
sviluppo e la funzione dell’orecchio interno. Mutazioni genetiche
interferiscono con passaggi critici nel suono di trasformazione, con
conseguente perdita dell’udito. Diverse mutazioni nello stesso gene
possono causare vari tipi di perdita dell’udito, e alcuni geni sono associati
con la sordità sia sindromica e non sindromica. In molte famiglie, il gene
(s) coinvolti devono ancora essere identificati.
La sordità
può anche derivare da fattori ambientali o una combinazione di genetica e ambientali fattori,
tra cui alcuni farmaci, infezioni peri-natale (infezioni che si verificano
prima o dopo la nascita), e l’esposizione a rumori forti per un periodo
prolungato.
Tipi
comprendono:
|
Gene |
Tipo |
|
|
DFNA1 |
||
|
DFNA2A |
||
|
DFNA2B |
||
|
DFNA3A |
||
|
DFNA3B |
||
|
DFNA4 |
||
|
DFNA5 |
||
|
DFNA8 / 12 |
||
|
DFNA9 |
||
|
DFNA10 |
||
|
DFNA11, neurosensoriale |
||
|
DFNA13 |
||
|
DFNA15 |
||
|
DFNA17 |
||
|
DFNA20 / 26 |
||
|
DFNA22 |
||
|
DFNA23 |
||
|
DFNA25 |
||
|
DFNA28 |
||
|
DFNA36 |
||
|
DFNA36, con dentinogenesi |
||
|
DFNA44 |
||
|
DFNA48 |
||
|
DFNA50 |
||
|
DFNB1A |
||
|
DFNB1B |
||
|
DFNB2, neurosensoriale (vedi anche sindrome di Usher) |
||
|
DFNB3 |
||
|
DFNB6 |
||
|
DFNB7 |
||
|
DFNB8, esordio infantile |
||
|
DFNB9 |
||
|
DFNB10, congenita |
||
|
DFNB12 |
||
|
DFNB16 |
||
|
DFNB18 |
||
|
DFNB21 |
||
|
DFNB22 |
||
|
DFNB23 |
||
|
DFNB24 |
||
|
DFNB25 |
||
|
DFNB28 |
||
|
DFNB30 |
||
|
DFNB31 |
||
|
DFNB35 |
||
|
DFNB36 |
||
|
DFNB37 |
||
|
DFNB39 |
||
|
DFNB49 |
||
|
DFNB53 |
||
|
DFNB59 |
||
|
DFNB63 |
||
|
DFNB67 |
||
|
DFNB77 |
||
|
DFNB79 |
||
|
DFNB84 |
||
|
DFNB91 |
||
|
DFNX1 |
||
|
DFNX2 |
Genetica
Sordità
sindromica può avere diversi modelli di ereditarietà. Tra il 75% e il 80%
dei casi sono ereditate in un modello autosomico recessivo, il che significa
che due copie del gene in ogni cellula sono alterati. Di solito, ciascun
genitore di un individuo con autosomica recessiva sordità è portatrice di una
copia del gene alterato. Questi vettori non hanno la perdita dell’udito.
Un altro 20%
al 25% dei casi di sordità non sindromica è autosomica dominante, il che
significa che una sola copia del gene alterato in ogni cella è sufficiente a
causare la perdita dell’udito. Le persone con autosomica dominante sordità
spesso ereditano una copia alterata del gene da un genitore che ha la perdita
dell’udito.
Tra l’1% e il
2% dei casi mostrano un modello X-linked di eredità, che significa che il gene
mutato responsabile per la condizione si trova sul cromosoma X. I maschi
con collegata a X sordità sindromica tendono a sviluppare sordità più grave in
precedenza in vita rispetto alle femmine che ereditano una copia della stessa
mutazione del gene. Padri non passeranno tratti X-linked ai propri figli,
poiché non passano sul cromosoma X alla prole maschile.
Sordità non
sindromica Mitocondriale, che risulta dalle modifiche al DNA nei mitocondri, si
verifica in meno di 1% dei casi negli Stati Uniti. Il DNA mitocondriale
alterato viene passato da una madre ai suoi figli e figlie. Questo tipo di
sordità non è ereditata dai padri.
sordità
progressiva ad esordio tardivo è la disabilità neurologica più comune degli
anziani. Anche se la perdita di decibel superiore a 25 è presente solo
l’1% dei giovani adulti di età compresa tra 18-24 anni di età tra, questo
aumenta al 10% nelle persone tra i 55-64 anni di età e circa il 50% negli ottantenni.
Il contributo
relativo dell’ereditarietà di deficit uditivo legata all’età non è nota,
tuttavia la maggior parte di sordità ereditaria ad esordio tardivo è autosomica
dominante e non sindromica (Van Camp et al., 1997). Oltre quaranta geni
associati con autosomica perdita di udito non sindromica dominante sono stati
localizzati e di questi quindici sono stati clonati.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Perdita
dell’udito Neurosensoriale Ereditaria Non Sindromica
Iris Schrijver
the journal of molecular diagnostics
November 2004Volume 6,
Issue 4, Pages 275–284
Schema dell’articolo
I.
Genetica
II.
Connessine
III.
Connexin
Funzione
IV.
Altri
geni
B.
COCH
(DFNA9)
C.
Mitocondriale
perdita dell’udito
E.
WFS1
(DFNA6, DFNA14, DFNA38)
V.
Universale
Newborn Hearing Screening
VI.
La
valutazione medica di perdita dell’udito
VII.
Test
genetici
VIII.
Considerazioni
etiche
IX.
Trattamento
X.
Conclusioni
XI.
Riferimenti
Enormi
progressi sono stati compiuti nella comprensione delle basi molecolari
dell’udito e perdita dell’udito.Attraverso i recenti progressi, abbiamo
cominciato a capire l’affascinante biologia del sistema uditivo e svelato nuovi
meccanismi molecolari di danni all’udito. Cambiamenti nella impatto
diagnostico dei test genetici sono verificati, nonché interessanti sviluppi in
opzioni terapeutiche. Diagnosi molecolare, che è già una realtà per
diversi geni acustici associati, sarà indubbiamente continuerà ad aumentare nel
prossimo futuro, sia in termini di numero di mutazioni testate e lo spettro di
geni. L’analisi genetica per la perdita dell’udito è usato soprattutto per
la diagnosi e il trattamento, e relativamente di rado per le decisioni
riproduttive, a differenza di altre malattie ereditarie. Ereditato perdita
dell’udito, invece, è caratterizzata da notevole eterogeneità
genetica. L’abbondanza di geni portano un gran numero di mutazioni, ma le
mutazioni specifiche in un singolo gene può causare la perdita dell’udito
sindromica o non sindromica.Alcune mutazioni predominano in singoli gruppi
etnici. Per diagnostici clinici e di laboratorio, è difficile tenere il
passo con le scoperte che si svolgevano. Questa recensione ha lo scopo di
fornire il quadro pertinente per diagnostici e un approccio pratico per
l’analisi di mutazione in non udenti.
La perdita dell’udito è comune a tutte le età. Si tratta di
un grave problema di salute pubblica perché colpisce 6-8% della popolazione nei
paesi sviluppati in cui tutte le cause sono combinati ed è il difetto congenito
più comune. 1 In
seguito all’attuazione dello screening uditivo neonatale universale (UNHS) è
stato trovato l’incidenza per essere ancora più alto di quanto si
pensasse. Circa uno su 1000 neonati sono sordi, uno in 300 bambini sono
colpiti con ipoacusia congenita di grado minore, e uno supplementare nel 1000
diventano profondamente udenti prima dell’età adulta. 2, 3 Prima della realizzazione di programmi di rilevamento e di
intervento precoce dell’udito, l’età media alla diagnosi era di 1,5 a 3 anni,
che è ben oltre l’inizio dell’intervallo critica per la voce e l’acquisizione
del linguaggio. 4 perdita
di udito non diagnosticate e ritardo diagnostico hanno un profondo impatto
sulla competenza linguistico-comunicativa, oltre che cognitivo e psicosociale
sviluppo. 5 riconoscimento
ritardata può portare all’isolamento più tardi nella vita. 6
La perdita dell’udito può essere dovuto a fattori ambientali,
difetti genetici, o una loro combinazione.Presbiacusia (perdita di udito legata
all’età) è generalmente considerato essere multi-fattoriale. Al contrario,
circa il 25% dell’infanzia udito deterioramento negli Stati Uniti è causato da
fattori ambientali come la prematurità, le infezioni, l’esposizione ai farmaci
ototossici, e traumi. Si stima che almeno il 50% di perdita di udito
prelinguale è causato da cambiamenti genetici, considerando l’eziologia rimane
oscuro nel restante 25%. La maggior parte di questi casi, tuttavia, si
presume essere di origine genetica. Così, cause genetiche rappresentano la
parte più consistente di tutti i casi di perdita dell’udito prelinguale. 7
La perdita dell’udito può essere classificato ulteriormente
diversi criteri, tra cui la gravità (lieve o da 20 a 39 dB, moderata o 40-69
dB, grave o 70-89 dB, e profonda o> 90 dB), 1 età dell’insorgenza (prelinguale o post-linguale), e
l’eziologia fisiologica. Ipoacusia trasmissiva è caratterizzata da
anomalie dell’orecchio esterno o anomalie degli ossicini dell’orecchio medio,
ipoacusia neurosensoriale è causa di un malfunzionamento dell’orecchio interno,
e la perdita dell’udito centrale è causata da difetti del nervo VIII, il tronco
cerebrale, o la corteccia cerebrale . La perdita dell’udito può essere
miscelato.
Ipoacusia genetica ha diverse eziologie e si stima che circa
l’1% di tutti i geni umani sono coinvolti nel processo di audizione. 8 Lasciando i modelli di eredità da parte, i conti perdita
dell’udito non sindromici per oltre il 70% di tutta la perdita dell’udito ereditaria. 9 sordità non sindromica o isolato non è associato con un
chiaro modello di altri difetti fisici. Al contrario, la sordità
sindromica è caratterizzata da manifestazioni aggiuntive, come la retinite
pigmentosa (ad esempio, la sindrome di Usher), gozzo eutiroideo e malformazioni
dell’orecchio interno (sindrome Pendred), dismorfismo cranio-facciale (sindrome
di Treacher-Collins), marfanoide habitus corpo (sindrome di Stickler), anomalie
renali (sindrome di Alport), o la presenza di lunghi intervalli QT (Jervell e
sindrome Lange-Nielsen). Sono state descritte diverse centinaia di
sindromi che coinvolgono la perdita dell’udito. 1 Sebbene utile quando caratteristiche sindromiche si possono
riconoscere, questa suddivisione pone un dilemma quando le manifestazioni
cliniche non sono completamente sviluppati.Ciò è particolarmente comune durante
l’infanzia, ma può anche essere causato da l’espressione genica
variabile. Ad esempio, il gozzo in autosomica recessiva sindrome di
Pendred può svilupparsi solo in età adulta, se non del tutto. In assenza
di altri sintomi evidenti all’esame obiettivo, la diagnosi di sindrome di
Pendred rischia di perdere.
Genetica
La perdita dell’udito può seguire un modello di autosomica
recessiva, autosomica dominante, X-linked, e l’ereditarietà
mitocondriale. La base genetica è molto complessa. Mutazioni
alleliche in alcuni geni possono causare la perdita recessiva e dominante
l’udito, le mutazioni nello stesso gene può causare sindromico o perdita di
udito non sindromica, e perdita di udito recessiva può essere causato da una
combinazione di due mutazioni in geni diversi dallo stesso gruppo funzionale.
Del minimo stimato del 50% dei casi con perdita uditiva
ereditaria, ~70% sono non sindromica e ~ 80% di questi sono autosomica
recessiva (10, Figura 1). Perdita di
udito non sindromica è molto spesso neurosensoriale. Può essere diviso in
DFNA (autosomica dominante sordità, ~15 al 20%), DFNB (autosomica recessiva
sordità, ~ 80%), DFN (sordità legata al cromosoma X, ~ 1%), e sordità
mitocondriale (almeno l’1%) .10, 11 autosomica dominante sordità
neurosensoriale (SNHL) spesso è post-linguale e progressiva, mentre SNHL
recessiva è prelinguale come una regola.

Figura
1
Una suddivisione di ipoacusia da causa (colonna 1),
presenza o assenza di funzioni associate nei casi ad eziologia
genetica (colonna 2), e modalità di trasmissione nel gruppo
non sindromica (colonna 3). Lescatole più piccole in colonna
3 rappresentano la perdita di udito legata al cromosoma X, che
rappresenta il ~ 1% di perdita di udito non sindromica, e perdita di udito
mitocondriale, che rappresenta almeno l’1%.DC, autosomica dominante; AR,
autosomica recessiva.
Immagine di grandi dimensioni | Visualizza Hi-Res Immagine | Scarica
PowerPoint Slide
{kind=link}
A causa della eterogeneità genetica enorme, l’identificazione di
geni e difetti del gene che influiscono sul processo di udienza è stato
impegnativo. Considerando la complessità del sistema uditivo, che richiede
l’interazione di una diversità di proteine compresi canali
ionici, matrice extracellulare, proteine citoscheletriche, e
fattori di trascrizione, questo non è sorprendente. Tuttavia, ostacola
l’identificazione di geni con metodi genetici tradizionali, come il
raggruppamento di più famiglie per analisi di linkage. Le analisi di
linkage è anche complicata dal fatto che più di una causa di perdita dell’udito
può separare in una famiglia dopo l’accoppiamento assortative, che è
relativamente comune tra gli individui sordi.
Studi genetici di perdita dell’udito hanno avuto successo in
popolazioni isolate e famiglie consanguinee.Anche dopo che il gene è stato
localizzato in una regione su un cromosoma, tuttavia, il processo di clonazione
posizionale tramite una mappa fisica seguita dall’identificazione trascrizione
può essere arduo. 12 Alcuni
loci identificati, compresi DFNB1, DFNA2 e DFNA3, sono stati trovati per porto
geni multipli. D’altra parte, alcuni geni hanno più assegnazioni locus
causa denominazioni iniziali errati. 13 identificazione
Gene è stato facilitato notevolmente dai recenti progressi nella genomica, la
libreria cocleare feto umano, e dalla ricerca in organismi modello come il
mouse. 12, 13
Nel campo della perdita di udito non sindromica, 21 geni
associati con ereditarietà autosomica recessiva, 20 associati con trasmissione
autosomica dominante, e uno con trasmissione recessiva legata al cromosoma X
sono stati identificati e caratterizzati (http://dnalab-www.uia.ac .be / dnalab / HHH /).
Connessine
Autosomica perdita di udito non sindromica recessiva nel locus
sul cromosoma 13q11-12 DFNB1 è caratterizzata da congenita, di solito non
progressiva, da lieve a insufficienza uditiva profonda. Il locus contiene
due geni, GJB2 e GJB6. GJB2 codifica
connexin 26, una proteina gap giunzione del gruppo beta con un peso molecolare
di 26 kD. Le connessine umani sono classificati per la loro massa
molecolare (riflessa dal numero nel nome connessina), e dalla portata di
identità di sequenza, che è indicato nei simboli gene per GJA, B, e C
sottotipi. I connexine sono molto simili e contengono loro regione
codificante in un singolo esone, separato dal 5′-UTR da un
introne. Identificazione della GJB2 gene è stato un punto
di riferimento nella genetica di perdita dell’udito, perché ha sottolineato il
ruolo centrale dei canali ionici gap junction cocleari. 14La sequenza codifica di GJB2 è
circondata interamente da esone 2 e si compone di 681 bp, che si traducono in
una proteina con 226 aminoacidi e comprendono il codone di stop. Esone 1 è
contenuta nel 5′-UTR. Le mutazioni in questo gene rappresentano la causa
più comune di perdita sporadica e autosomica recessiva non sindromica
neurosensoriale dell’udito (SNHL). Essi sono responsabili per circa la
metà dei casi negli Stati Uniti, Europa, Australia e Israele, e sono stati
segnalati in altre popolazioni pure. 15
La mutazione più comune è una delezione di una singola guanina
da una stringa di sei (35delG). Spiega questa mutazione per più di due
terzi delle mutazioni e dei risultati identificate in un frame-shift con
premature terminazione della proteina. Rimane ancora da determinare perché
questa mutazione ha una frequenza relativamente alta, ma è stato suggerito che
35delG si trova in una regione hypermutable. 16 Sia un consenso motivo Chi locale
e una sequenza TGGGG, che sono stati collegati a beta-globina mutazioni e la
ricombinazione dei geni immunogobulin, potrebbero svolgere un ruolo. Se
slittamento e mispairing dei fili durante la sintesi del DNA determinano
l’elevata incidenza di questa mutazione, tuttavia, etnica dovrebbe non poco
contribuire variazione nella frequenza. Tuttavia, la prevalenza di 35delG
sembra variare notevolmente tra le popolazioni. Un’alternativa alla mutazione
punto caldo ipotesi viene proposta da Van Laer et al, 17 il quale propone che l’alta
frequenza di questa variante risultati da un comune fondatore. Perché la
mutazione è pensato per essere evolutivamente antico, aplotipo sharing è
osservata solo in una piccola regione cromosomica. Anche se queste ipotesi
sembrano essere contraddittorie, entrambi i fenomeni possano aver contribuito
alla elevata frequenza di alleli di questo singola mutazione. 17 La frequenza complessiva portante
della 35delG negli Stati Uniti raggiunge il 2,5% in alcuni studi, ma sembra
variare in base alla popolazione.17, 18, 19
. La mutazione 167delT è la mutazione più comunemente
identificato nella popolazione ebraica ashkenazita, dove ha una frequenza
portante di circa il 4% 20 nel
sud-est asiatici, 235delC è la più diffusa con una frequenza portante di ~ 1:
100. 21 un
elenco di tutte le mutazioni pubblicati così come polimorfismi comuni inGJB2,
è disponibile per la correlazione clinica e l’interpretazione alla home page di
connessina-sordità(http://www.crg.es/deafness/). Circa 110 i singoli
non sindromici GJB2 sono state descritte varianti del gene:
otto sono dominanti, 89 sono recessive, e 11 sono di significato
sconosciuto. Mutazioni riportate sono sciocchezze, missense, tranciati, e
le mutazioni frame-shift così come in-frame delezioni(http://www.crg.es/deafness/).
Anche se fino a metà degli individui con autosomica recessiva
SNHL avere GJB2 mutazioni, ~ 10 al 50% portare solo uno. 15 Un ruolo per GJB6, il
gene adiacente a GJB2 sul cromosoma 13, è stata suggerita nel 1999,
quando una mutazione dominante ( T5M) è stata descritta. 22 La mutazione più comune in GJB6,
però, è una delezione> 300 kb che causa SNHL non sindromica quando
omozigote, o se è presente sulla allele opposto di un GJB2 mutazione. 23 GJB2 e GJB6 sono
solo ~ 35 kb a parte. GJB2 si trova sul lato centromerico(Figura 2). GJB6 è
molto simile a GJB2, ma non interrotto da introni. 22 Entrambi i geni sono espressi
nella coclea dove possono combinarsi per formare hemichannels più unità nella
cella membrana, e la funzione come parte integrante del regolamento di potassio
nell’orecchio interno. Un modello di topo con connessina 30 carenza
dimostra profonda perdita dell’udito congenito. Un meccanismo DIGENICA di
eredità, di conseguenza, è plausibile. In alternativa, la grande
eliminazione potrebbe influenzare un elemento normativo monte da scoprire
di GJB2. 24

Figura
2
Rappresentazione schematica del braccio lungo del cromosoma 13,
12 bande (non in scala). I GJB2 eGJB6 geni,
che codificano connessina 26 e connessina 30, rispettivamente, sono
adiacenti. Δ, un 309-kb 15delezione,
che elimina una parte del GJB6 gene ed è associata con perdita
di udito in forma omozigote recessivo, o in stato eterozigote, in combinazione
con una mutazione nel GJB2. Questa delezione era precedentemente
pensato per estendersi 342 kb.
Immagine di grandi dimensioni | Visualizza Hi-Res Immagine | Scarica
PowerPoint Slide
{kind=link}
Come si è visto con la mutazione 35delG nel gene connessina 26,
la prevalenza della delezione del gene connessina 30 sembra variare
notevolmente a seconda etnia. La frequenza delezione in materie di studio
con uno o zero GJB2 mutazioni è più alta in Israele (71,4%),
mentre il GJB6 mutazione avviene in circa il 20% della
popolazione degli Stati Uniti con problemi di udito. 15 Il GJB6 eliminazione
può spiegare ~ 10% di tutto DFNB1 alleli con una gamma estremamente ampia
basata sull’origine etnica. Finora, l’informazione è stime molto limitate
e affidabili preclusi da un piccolo numero di campioni in alcuni
paesi. Anche all’interno degli Stati Uniti, ulteriori studi saranno
necessari come la frequenza complessiva può variare notevolmente da stato a
stato.Tuttavia, connessina 30 contribuisce in modo significativo la base
molecolare di perdita dell’udito. Oltre ad un effetto diretto, è possibile
che il GJB6 delezione modifica il fenotipo in individui che
portano due recessiveGJB2 mutazioni. Non ci può essere
notevole variabilità intra-familiare con perdite che vanno da lievi a profonde
udito. Finora, tuttavia, ciò non è stato esplorato. 25
Non sindromica sordità dominante associata a GJB2 mutazioni
è insorgenza precoce, da moderata a grave, e (a differenza di autosomica
recessiva GJB2 sordità legati) tipicamente
progressive. Dominanti GJB2 mutazioni, però, spesso hanno
effetti pleiotropici. Disturbi dell’udito è stata riportata in
associazione con disturbi della pelle tra palmoplantar keratorderma (PPK, la
sindrome di Vohwinkel), cheratite-ittiosi-sordità, e sindromi ittiosi sordità
hystrix-like. 26 GJB6 è
stata anche associata con la sindrome di Clouston (hydrotic displasia
ectodermica), che è autosomica dominante e può verificarsi con la sordità. 27, 28
Connexin Funzione
Più di 20 proteine connessina ampiamente espresse
sono state identificate finora(http://www.ncbi.nlm.nih.gov/). Attraverso
la formazione di giunzioni, consentono la comunicazione tra cellule adiacenti e
sono coinvolti in numerose funzioni cellulari tra cui la crescita cellulare, la
differenziazione, la reazione a segnali, la sincronizzazione di attività in
tessuti eccitabili, e omeostasi. 29 I
livelli di espressione differenti connexin isotipi sono interconnesse, come è
stato dimostrato in un mouse connessina 32 knock-out, che ha avuto la riduzione
simultanea di connessina 26 espressione in epatociti. 30 connessine 26, 30, 31, 32, e 43
sono espressi nell’orecchio interno se soddisfano un importante ruolo nella
udito normale.
Nella coclea, la endolinfa ricco di potassio, si mette in moto
ondoso dalla vibrazione meccanica del suono.Questo devia microvilli sugli apici
cellulari e sposta l’stereocilia-actina contenenti associato. Potassio fluisce
poi nelle cellule cigliate e depolarizza la membrana risultante in un
potenziale d’azione, che attiva il nervo acustico. Per mantenere il
gradiente di potassio elettrochimica tra le cellule dei capelli e la endolinfa,
però, gli ioni potassio devono essere riciclati dalle cellule dei capelli di
nuovo al endolinfa. 31 Anche
se la loro esatta funzione e il ruolo nella perdita di udito rimangono
sconosciute allo stato attuale, è stato messo in avanti che giunzioni connexin,
attraverso la comunicazione intercellulare, influenzano l’ambiente ionico delle
cellule epiteliali cocleari e potassio ricircolo. 32 trova nella membrana cellulare,
sei connessine dello stesso tipo o differenti tipi formare una connessone, che
crea un poro nella membrana e canale di comunicazione intercellulare quando
allineato con una seconda connessone in una cella adiacente 33 (Figura 3). Riduzione
di valore delle normali risultati di funzione dei canali ionici a morte delle
cellule dei capelli e la sordità permanente. 32, 34, 35

Figura
3
Sei proteine connessine (cxi) gruppo per formare
un hemichannel nella membrana cellulare. Questo canale è chiamato
connessone (CXO). Connexons possono essere fatte di sei connessine
identici (omomerici) o di tipo diverso (eteromerico). Due connexons vicini,
che creano un canale di comunicazione funzionale, possono essere identici
(omotipica, come mostrato sotto A) o di diversa composizione
(eterotipica, come mostrato sotto B).
Immagine di grandi dimensioni | Visualizza Hi-Res Immagine | Scarica
PowerPoint Slide
{kind=link}
Gli studi sugli animali e in vitro di ricerca
dei singoli connessine umani hanno illuminato le diverse, selettivi, e
complessi compiti nei singoli tessuti. La molteplicità di funzioni
cellulari è facilitata dalla espressione di una varietà di connessine in ogni
tipo di cellula. Attraverso combinazione di diversi connessine come
elementi costitutivi dei singoli connexons, e attraverso la formazione di
diverse coppie di connessone, questi canali ionici dimostrano grande varietà
nelle loro funzioni. D’altro canto, la struttura connessone può limitare
alcune interazioni cellula-cellula per incompatibilità selettiva. 29
Connessine Mutant espressi in coppie di Xenopus ovociti,
con quantificazione dell’attività canale tra le due cellule mediante la tecnica
del doppio morsetto di tensione hanno dimostrato una riduzione o completa
mancanza di funzione connessone. Considerando che la perdita di funzione è
il principale meccanismo patogenetico per mutazioni recessive connessine,
mutazioni dominanti esercitano un effetto dominante negativo sulla proteina
wild-type. L’inibizione negativamente dominante è molto probabilmente il
risultato della interazione diretta di connessine normali e difettosi sulla membrana
cellulare. 22 Non
solo si verifica per effetto di un connexin difettoso su un intatto uno dello
stesso tipo, ma può anche essere tra- dominante, come è stato
dimostrato per la connessina 26 e 43, che può co-localizzare. 36
Connessine 26 e 30 sono co-espressi nell’orecchio
interno. Connessina 30 knock-out così come i modelli di topo in cui
connessina 26 è stata selettivamente ablato nell’orecchio interno (un approccio
scelto perché connessina 26 – / – mice non sono vitali) hanno grave ipoacusia,
suggerendo che queste due connessine sono in grado di si completano a vicenda
nella parte interna dell’orecchio. 37 Una
singola mutazione recessiva in ogni gene può anche portare a SNHL non
sindromica attraverso un meccanismo apparentemente DIGENICA.Il deficit uditivo
potrebbe essere dovuto ad un effetto di dosaggio, ma si può prevedere che la
allele wild-type di ogni gene sarebbe in grado di compensare. Considerando
che ci sono sei connessine in ogni connessone, tuttavia, è probabile che le
proteine anomale sono incorporati nella maggioranza dei connexons
heteromeric e omomerici. Questo si traduce poi in connexons difettosi che
non possono ricircolare efficace di potassio, sono privi di potenziale
elettrico, e non riescono a fornire un ambiente in cui le cellule ciliate
sopravvivere. Finora, tuttavia, la base funzionale dell’osservazione
DIGENICA non è chiarita.
Altri geni
Diverse centinaia di geni sono coinvolti nella complessa
biologia dell’udito. Piuttosto che alla lista tutti i geni completamente
caratterizzati, la seguente sezione si propone di rivedere i geni diversi GJB2 e GJB6 coinvolti
nella perdita di udito non sindromica per i quali le prove di laboratorio
clinico è attualmente disponibile(http://www.genetests.org/servlet/access ) (Tabella 1).
|
Tabella 1geni coinvolti nella non sindromica |
||||||
|
Chrom.posizione |
Locus / mutazione |
Simbolo Gene |
In lei. |
Proteina |
Funzione |
Rif. |
|
13q11-12 |
DFNB1 |
GJB2 |
AR |
Connexin |
Gap |
|
|
13q12 |
GJB6 |
AR |
Connexin |
Gap |
||
|
7q31 |
DFNB4 |
SLC26A4 |
AR |
Pendrin |
Trasportatore |
|
|
14q12-13 |
DFNA9 |
COCH |
ANNO |
Cochlin |
Proteina |
|
|
Mitoch. |
1555A> |
MTRNR1 |
Mito. |
12SrRNA |
||
|
7445A> |
MTTS1 |
tRNA |
||||
|
7472insC |
||||||
|
7511T> |
||||||
|
Xq21.1 |
DFN3 |
POU3F4 |
XL |
Pou |
Fattore |
|
|
4p16.1 |
DFNA6 |
WFS1 |
ANNO |
Wolframin |
Non |
|
Chrom, cromosomica; Inher, eredità.; Mitoch,
mitocondriale.; Ref., Riferimenti.
SLC26A4 (DFNB4)
Le mutazioni in SLC26A4 sono associati con
autosomica recessiva SNHL e con la sindrome di Pendred. 38, 39Questa è una delle forme più comuni di
sordità sindromica, ma ha riconosciuto sotto-probabile. Si è associato con
una coclea dismorfico che contiene 1,5 invece di 2,5 giri (Mondini displasia) e
con l’allargamento l’acquedotto vestibolare, che può essere esaminato da
tomografia computerizzata. Il deficit uditivo è in genere grave e spesso
stabile, anche se può essere progressiva. Il SLC26A4 gene
codifica Pendrin, cloruro / trasportatore di ioduro. Ulteriori
manifestazioni della sindrome di Pendred includono gozzo eutiroideo, che deriva
dalla limitata capacità della tiroide di iodio durante la organify ormone
tiroideo biosintesi, ma questa caratteristica non è né specifica né
sensibile. Gozzo può essere un sintomo clinico di altri disturbi, e non è
sempre presente nella sindrome di Pendred causa di penetranza
incompleta. Anche quando espresso, spesso non si manifesta fino all’età
adulta. Un test di laboratorio che può essere utile è il test di scarico
perclorato, che valuta la organificazione di ioduro. Test molecolare è
prezioso e fattibile in questo disturbo, come quattro ricorrenti mutazioni
spiegano ~75% dei cromosomi colpiti. Le mutazioni possono tuttavia essere
trovati in tutto il gene. 40
COCH (DFNA9)
Il COCH gene codifica Cochlin, una proteina
abbondantemente espressa nella matrice extracellulare dell’orecchio
interno. La perdita dell’udito associati a difetti in questo gene è in
genere autosomica dominante, non sindromica, post-linguale con un esordio in
età adulta, e progressiva. Gli individui affetti hanno l’unico osso
temporale scoperta istopatologica di deposizioni mucopolisaccaridi, che
sembrano soffocare le fibre dendritiche. Sintomi vestibolari quali
squilibrio possono essere presenti. Questa malattia ha somiglianze
cliniche per la malattia di Meniere, ma, al contrario, mostra la perdita
dell’udito ad alta frequenza. 41
Mitocondriale perdita dell’udito
SNHL è presente nel 42-70% dei soggetti con disturbi
mitocondriali e può essere non sindromica o sindromica. Mutazioni del DNA
mitocondriale sono state identificate in> 3% dei pazienti con
SNHL. Questa cifra è destinata ad aumentare a causa di un previsto aumento
dei test genetici e la consapevolezza. 42mitocondriali
le mutazioni di perdita dell’udito sono trasmesse esclusivamente attraverso la
linea materna, e dimostrano completa o quasi completa omoplasmia, il che
significa che la mutazione è presente in (quasi) tutto dei genomi mitocondriali
all’interno di un individuo. 43
Fino al 25% dei pazienti che ricevono aminoglicosidi esperienza
SNHL, anche quando somministrato a livelli terapeutici e per un periodo di
tempo breve. Il cinquanta per cento delle persone colpite portano i 12S
ribosomiale RNA mutazione 1555A> G nel MTRNR1. SNHL potrebbe
essere evitato in molti pazienti se l’analisi del mtDNA verrebbe eseguita di
routine e un alto indice di sospetto erano presenti prima della
somministrazione aminoglicosidi. 42 Questa
mutazione è anche una causa indipendente di non sindromica, e generalmente più
mite, la perdita dell’udito in molteplici etnie.
La mutazione 7445A> G nel gene che codifica la serina tRNA
provoca perdita dell’udito, ma penetranza dipende sul aplotipo mitocondriale in
singole popolazioni. Ciò ha portato ad ipotizzare che sia modificatori
genetici accessori o fattori ambientali giocano un ruolo nell’espressione di
perdita dell’udito. Alcune mutazioni, come un inserimento citosina in
posizione 7472 nel gene tRNASer, sono associate a sordità neurosensoriale
progressiva combinata con manifestazioni neurologiche. La mutazione
7511T> C in tRNASer non è associato con manifestazioni neurologiche, ma
porta alla perdita progressiva dell’udito con l’età variabile di esordio. 44più mutazioni sono state identificate
in pazienti con SNHL non sindromica, ma questi sono ancora in corso di
valutazione per patogenicità, 43 (http : //dnalab-www.uia.ac.be/dnalab/hhh/).
Mitocondriale SNHL può essere sindromica e può essere associata
a mutazioni puntiformi (compresi quelli in tRNALys e tRNASer), delezioni,
duplicazioni e (http://dnalab-www.uia.ac.be/dnalab/hhh/). Sindromi
associate con perdita uditiva mitocondriale includono quelli causati dalla 3243A>
G mutazione puntiforme nel MTTL1, il gene che codifica per la leucina
tRNA. In questi casi, la perdita dell’udito esiste in concomitanza con
la sindrome di Kearns-Sayre, encefalopatia mitocondriale con acidosi lattica e
episodi di simil-ictus (MELAS), diabete ereditarietà materna e la sordità, o
oftalmoplegia esterna progressiva cronica.
La patogenesi della perdita uditiva mitocondriale è dovuta
all’elevata adenosina trifosfato (ATP) requisito nelle cellule ciliate
cocleari. Una riduzione di ATP disponibile, causata da una disfunzione
della fosforilazione ossidativa mitocondriale causa di mutazioni, provoca
disturbi del gradiente ionico nell’orecchio interno. Così, mutazioni
mitocondriali possono contribuire alla perdita progressiva dell’udito di invecchiamento,
presbiacusia. 42
POU3F4 (DFN3)
POU3F4 codifica per un fattore di trascrizione (POE è
l’abbreviazione di Pit, ottobre, e il DNA Unc domini di legame). 45 mutazioni sono state trovate
disperse attraverso il gene, con il clustering nei domini POU. Sono
causale nella X-linked, non sindromica, progressiva e profonda ipoacusia
neurosensoriale (DFN3), che possono dare prova di una componente conduttivo a
causa della fissazione staffa. I staffa è un osso a forma di staffa
nell’orecchio medio. POU3F4 correlate perdita dell’udito è un
altro disturbo per il quale l’imaging studi di TC possono essere utili, come un
allargamento del canale acustico interno, e una dilatazione al confine tra la
acustico interno canale e l’orecchio interno può essere visto. Come
risultato, la pressione è aumentata e perilinfatica perilinfa dall’orecchio
interno può trasmettere durante la rimozione chirurgica della staffa.
WFS1 (DFNA6, DFNA14, DFNA38)
WFS1 è stato descritto nel 1998 da Strom ed altri 46 e più di 90 sono state
identificate mutazioni nel WFS1gene da allora. WFS1 codifica
l’wolframin glicoproteina ed è associata con autosomica recessiva sindrome di
Wolfram in presenza di mutazioni inattivanti. Esso è legato anche a un
aumento della suscettibilità al suicidio e malattia mentale, così come il
diabete. Non mutazioni inattivanti, con un effetto dominante negativo
presunto, sono una causa comune di autosomica dominante SNHL bassa frequenza
(DFNA6 / 14/38). 47 La
maggior parte di queste mutazioni si trovano nel dominio della proteina
carbossi-terminale. La perdita dell’udito mostra tipicamente esordio
nell’infanzia ed è progressiva, ma non si avanza alla sordità profonda. La
maggior parte dei pazienti non hanno alcuna significativa compromissione
dell’udito nelle frequenze importanti per il discorso. Così, la maggior
parte non richiedono apparecchi acustici. 48
Universale Newborn Hearing Screening
In passato, un approccio di screening con l’inserimento di soli
bambini ad alto rischio non è riuscito a identificare almeno il 50% dei bambini
con perdita di udito, e molti neonati senza fattori di rischio è rimasta non
diagnosticata solo dopo 18 mesi di età. Se la diagnosi e l’intervento si
svolgono prima dei 6 mesi di età, tuttavia, un livello quasi adeguato all’età
delle competenze linguistiche può essere realizzato. Nel 2000, il Comitato
misto per l’Audizione Infant ha riconosciuto universale Newborn Hearing
Screening (UNHS). 49 Lo
scopo è quello di fornire lo screening a tutti i neonati sentire prima dell’età
di 1 mese, con la conferma della perdita di udito nei bambini che non superano
il primo , o una proiezione successiva, attraverso una valutazione audiologica
dall’età di 3 mesi. Trattamento completo può quindi essere iniziata prima
dell’età di 6 mesi. 6 La
maggior parte dei bambini identificati attraverso questo programma hanno
genitori con udito normale.
La perdita dell’udito può essere identificato da diversi metodi complementari
diversi. Seguendo le linee guida dal National Institutes of Health, tutti
gli stati degli USA hanno adottato UNHS, ma algoritmi e di collaudo di
variare. I due metodi principali utilizzati per il controllo neonatale
sono emissioni otoacustiche e uditivi del tronco encefalico risposta
automatica. 2
Una limitazione di UNHS è che non tutti i casi di perdita
dell’udito dell’infanzia verranno rilevati. Lo screening neonatale
dell’udito potrebbe non riuscire a identificare i bambini con perdita
progressiva dell’udito, che rappresenta circa il 15% dei bambini in età
prescolare con SNHL. 11 La
progressione è stata anche descritta nella perdita dell’udito prelinguale
solitamente stabile associata a mutazioni GJB2. In due bambini con
mutazioni omozigote 35delG, la perdita dell’udito sembra essere stata
prelinguale ma non congenito. Entrambi i bambini superato test dell’udito
neonato di risposta uditivi del tronco encefalico e di un campo audiogramma
gratuito, rispettivamente. Ciò implica che un bambino può passare i UNHS
ma potrebbe ancora diventare gravemente colpiti primi anni di vita. 50
Il test UNHS è solo il primo passo di un programma di successo e
conveniente. L’obiettivo principale è la diagnosi precoce e la gestione,
lo sviluppo del linguaggio normale, e il successo a lungo termine dopo
l’intervento. Gestione dei dati e il monitoraggio dei bambini per il
follow-up sono attualmente in fase di sviluppo. Finora, tuttavia, non vi è
alcun protocollo universale per la gestione medica dei bambini non udenti.
La valutazione medica di perdita dell’udito
La valutazione medica deve iniziare non appena la perdita dell’udito
è sospettato di un prenatale, medico, e la storia completa della
famiglia. I fattori di rischio quali bassi punteggi di Apgar, basso peso
alla nascita, sofferenza respiratoria, ventilazione meccanica, l’ammissione in
terapia intensiva neonatale, iperbilirubinemia, displasia retrolental, anomalie
cranio-facciali e anomalie cromosomiche dovrebbero essere valutate in questo
momento. 6
L’esame clinico e genetico sono necessari per escludere le
caratteristiche spesso sottili compatibili con una eziologia infettiva
sindromico o congenite. Inoltre, un esame oftalmologico devono essere
effettuati esclusivamente a causa anomalie oculari sono presenti in più della
metà dei bambini con grave di sordità profonda. 51 Ogni paziente con perdita di
udito inspiegabile dovrebbe anche avere una ecografia renale e neuro-imaging
dell’osso temporale. Studi di imaging possono includere risoluzione
elevata TAC per valutare la presenza di una malformazione Mondini, o una
risonanza magnetica per visualizzare il nervo acustico, escludere aplasia, e di
escludere infettiva distruzione dell’orecchio interno. Una risonanza
magnetica è particolarmente importante prima di un intervento chirurgico di
impianto cocleare.
Gli esami di laboratorio deve essere personalizzata e diretta
verso il sospetto diagnostico. I test possono includere un anticorpo test
IgM nei primi anni di vita per valutare la possibilità di infezione
intrauterina, test per emoglobinopatie come questi possono essere associati a
SNHL, analisi delle urine e test di funzionalità renale nei bambini con
possibile sindrome di Alport, test della tiroide di governare fuori una
carenza, TSH e un test di scarica perclorato in sospetta sindrome Pendred, e
una valutazione di disturbi metabolici come le malattie da accumulo
lisosomiale. 11 un
ECG per valutare l’intervallo QT devono essere effettuate se Jervell e
Lange-Nielsen o sindrome di Romano-Ward sono sospetto. Qualsiasi valutazione
della perdita di udito richiede un approccio multidisciplinare, che dovrebbe
includere la consulenza e il supporto per i genitori. Un recente sondaggio
ha rivelato che l’identificazione della causa della perdita dell’udito è la
priorità più alta di genitori che imparano che il loro bambino è non
udenti. 6
Test genetici
GJB2 (connessina 26) L’analisi dovrebbe essere il primo passo
nella analisi di mutazione per non sindromica sordità neurosensoriale, in
quanto è la causa più comune nella sua categoria. I campioni utilizzati
per le prove possono includere sangue e nei tessuti periferici, ma anche i
campioni meno invasive come le cellule buccali ottenute con un tampone, o parte
delle macchie di sangue raccolti per lo screening neonatale. Entrambe le
modalità di raccolta di DNA ad alleviare l’obbligo di eseguire la venipuntura
su bambini molto piccoli. GJB2mutazioni possono essere identificati
con una varietà di test allele-specifici, tra cui PCR seguiti da restrizione
digestione, PCR con allele-specifico ibridazione, primer extension, o in tempo
reale PCR. Tali metodi sono rapidi, economico e altamente sensibile e
specifico ma limitando perché il numero di mutazioni puntiformi indagato è
tipicamente bassa. Come osservato in numerose altre condizioni, origine
etnica svolge un ruolo importante nella prevalenza e frequenze di mutazioni
come illustrato dalle mutazioni più comunemente osservati negli ebrei Ashkenazi
(167delT), asiatici (235delC) e caucasici (35delG). La mutazione 35delG è
nettamente raro negli afro-americani, ma la prevalenza di SNHL non lo
è. Molto probabilmente, altri GJB2 alleli quali R143W
giocano un ruolo. Questa mutazione è stata rilevata nella maggior parte
dei soggetti dello studio non udenti in Ghana. 52 Così, origine etnica dovrebbe
essere un fattore nel decidere quale tipo di test è la più appropriata per un
paziente.
Metodi di scansione Mutazione quali DGGE, TTGE, SSCP, DHPLC e
hanno bisogno di essere completamente ottimizzato per creare risultati
riproducibili. A seconda della tecnica, condizioni, e livello di
ottimizzazione, alcuni di questi metodi sono uno schermo generale mutazione con
cui alcune mutazioni possono perdere. Il sequenziamento è il metodo più
completo e definitivo, come quasi tutte le mutazioni puntiformi, nonché piccole
delezioni e inserzioni possono essere rilevati. Le dimensioni
relativamente ridotte della GJB2 gene rende adatto per questo
approccio. Quasi tutte le mutazioni sono stati rilevati nella regione
codificante, che è interamente comprendeva dalla seconda esone. Tuttavia,
poiché la maggior parte dei laboratori si concentrano solo su questo settore,
non è completamente risolto se l’inclusione dell’esone uno sarebbe
vantaggioso. 52 Inconvenienti
di sequenziamento del DNA diretta sono l’incapacità di rilevare delezioni di
interi esoni o geni e l’interpretazione alta intensità di lavoro, anche se la
sequenza software confronto facilita questo compito. Analisi di sequenza
in presenza di una mutazione frame-shift può essere difficile perché altre
mutazioni sottostanti possono essere presenti. Se viene rilevata una
mutazione frame-shift, primers dovrebbero essere utilizzate per interrogare la
sequenza in entrambe le direzioni. 53 Grandi
delezioni e inserzioni possono essere studiate con un lungo approccio PCR,
accoppiato con l’analisi a base di gel per il dimensionamento.
Il GJB6 eliminazione dovrebbe essere esaminato
in una seconda fase in individui in cui nessuno, o solo un singolo, GJB2 stata
rilevata mutazione. 54 Ciò
può, per esempio, essere ottenuto mediante l’uso di primer che fiancheggiano il
grande delezione, in quanto questo è un ricorrente cancellazione con gli stessi
punti di interruzione. Solo individui con delezione dimostrerebbero
presenza del prodotto, mentre un prodotto di controllo esterno della
soppressione potrebbe essere utilizzato per la verifica di
amplificazione. 23 L’GJB2 e
sei prodotti possono essere anche mirata simultaneamente da una PCR multiplex
per valutare la presenza di eliminazione dal gel di agarosio elettroforesi,
seguita da sequenziamento del DNA diretto della griglia di lettura aperta
nel GJB2 gene. 53
Oltre a UNHS e un controllo medico generale, potenti argomenti
possono essere fatte per i test genetici in pazienti con perdita di
udito. 1) Nella maggior parte degli individui con ipoacusia genetica, una
eziologia non è possibile altra via, perché gli studi di imaging sono negativi,
caratteristiche extra-uditiva non sono evidenti, e il fenotipo perdita di udito
non consente la categorizzazione. La notevole e continuo progresso nella
nostra comprensione della patologia molecolare di ipoacusia, al contrario,
consente sempre l’individuazione di un difetto genetico specifico. 2)
Poiché l’analisi molecolare è essenzialmente non-invasive, sedazione o anestesia
generale di neonati e bambini possono essere evitabili e la necessità di una
più ampia, e costoso, le prove possono essere ridotti. 3) L’analisi
molecolare può risultare in una diagnosi accurata e precoce, che facilita lo
sviluppo cognitivo ottimale. 4) L’analisi molecolare può essere utile per
la diagnosi di sordità sindromica prima funzionalità aggiuntive emergere (ad
esempio, nella sindrome di Pendred o Jervell e Lange-Nielsen sindrome), e in
grado di distinguere gli individui con mutazioni mitocondriali che sono a
rischio per la perdita dell’udito iatrogena se trattati con
aminoglicosidi. I rapporti pubblicati sono, in generale, esclusi i casi
con evidenti fattori di rischio ambientali o sindromiche pazienti di perdita
dell’udito da GJB2 analisi. Dal momento che molti
individui con questi fattori di rischio e la perdita dell’udito hanno
dimostrato di portare GJB2mutazioni dopo tutto, però, i test
genetici può essere opportuno in questi casi. 5) GJB2 varianti
possono influenzare l’espressione di perdita dell’udito sindromica, ambientale,
progressivo, e all’età. Perché GJB2vettori hanno ridotto la
funzione delle cellule dei capelli, le mutazioni eterozigoti possono
contribuire a manifestazioni più gravi o precedenti. 52, 55 6) Altri benefici includono la
conoscenza associata del modello di eredità e di consulenza genetica più
accurata.
Come saranno disponibili ulteriori dati, correlazioni
genotipo-fenotipo possono essere fatte e il significato clinico delle singole
mutazioni, o loro combinazioni, può essere valutata più affidabile. La
domanda per i test molecolari è già in aumento con la scoperta dei difetti
molecolari alla base varie perdita dell’udito. Lo spettro di test
molecolari disponibili clinicamente o su una base di ricerca è in crescita e
può essere seguito sul sito web
GeneTests (http://www.genetests.org/servlet/access). E c’è da
aspettarsi che i test genetici diventerà parte integrante di ascoltare le
valutazioni di perdita a tutte le età, e che UNHS saranno accoppiati con le
linee guida per il follow-up. Anche se una mutazione non può essere
identificato al momento, ri-contattare e test genetici di follow-up può essere
giustificata quando vengono sviluppati saggi molecolari supplementari.
Risultati diagnostici molecolari dovrebbero sempre essere
interpretati con cautela, in quanto la nostra conoscenza delle basi molecolari
di perdita dell’udito è ancora in evoluzione. Questo pone una sfida per
gli operatori sanitari, come pochi sono specializzati in questo settore. Una
errata percezione comune è che una mutazione screen negativo esclude la sordità
genetica. I nostri rapporti e interpretazioni deve essere personalizzata,
dettagliate, e riflettono incertezze attuali conoscenze al momento opportuno. Patogenicità
non è ancora stato determinato per tutte le mutazioni descritte (vedi la
homepage connessina-sordità per i dati connessina attuali, http://www.crg.es/deafness/)
e nuove mutazioni sono scoperti da approcci globali come il sequenziamento del
gene. In tali casi, la
patogenicità può dedurre dalla conservazione amminoacidica tra le specie, luogo
della mutazione ad un residuo corrispondente a mutazioni patogene in geni
correlati, impatto di una mutazione in carica e la polarità, coesistenza con
altre mutazioni nello stesso gene o in un altro, tipo di perdita dell’udito
identificato, studi familiari, e l’assenza della nuova mutazione in un grande
gruppo di soggetti di controllo con lo stesso sfondo etnico. Anche così,
la patogenicità può essere risolto solo con espressione e studi
funzionali. 53 Cautela
deve essere praticata anche in termini di eredità, come autosomica dominante e
perdita di udito recessivo può essere causata da mutazioni nello stesso
gene. Correlazioni genotipo-fenotipo stanno solo ora emergendo e sono
oggetto di studio su scala più ampia per determinare l’effetto di composti set
mutazione eterozigoti. Anche con mutazioni ben noti, come la mutazione
35delG nel GJB2 gene, tuttavia, la variabilità intra-familiare
da lieve a sordità profonda può essere osservato e la prognosi accurata non è
attualmente possibile. Ci possono essere mutazioni missense specifiche che
esercitano un’influenza nello stato eterozigote. Mutazioni eterozigoti
possono anche influenzare la suscettibilità alla presbiacusia o menomazione
dell’udito causata dal rumore attraverso un effetto semi-dominante. 20
Considerazioni etiche
I test genetici per la perdita di udito e la sordità non sono
collettivamente percepito come vantaggiosa.Soprattutto nella comunità dei non
udenti in generale, la sordità è né considerato negativo, né limitando.Questa
comunità ha una propria cultura linguistica (linguaggio dei segni), i valori e
l’identità di cui la sordità è parte integrante. Non è percepita come una
condizione medica. Di conseguenza, i progressi nella ricerca di perdita
dell’udito e test genetici potrebbero essere percepite come dannose. Servizi
genetici possono essere considerati, tuttavia, perché alcuni individui
preferiscono avere bambini sordi. Servizi di consulenza genetica per le
famiglie con sordità possono essere solo efficace e appropriato se si prendono
i valori sociali della comunità dei non udenti in considerazione. 56
Per sentire i genitori, d’altra parte, avere un figlio non
udente o sordo solleva tipicamente molte domande e preoccupazioni per il
futuro. La possibilità di determinare l’eziologia della perdita uditiva
del loro bambino attraverso metodi non invasivi, la prospettiva di un
trattamento mirato e cura completa primi anni di vita, insieme con una
comprensione della modalità di trasmissione e del caso di recidiva, sono generalmente
accolti e portare ad una maggiore l’accettazione nella comunità
udienza.&nbsnbsp;Non a caso, la diagnosi prenatale sarebbe considerato ascoltando
individui più di due volte più spesso di quanto da persone sorde (49% contro il
21%), con l’audizione con problemi di caduta tra (39%). All’interno di
tutti questi gruppi, l’interruzione della gravidanza sarebbe considerato solo
da una piccola minoranza. 57
Atteggiamenti generali del più ampio dell’udienza, sordo, e la
comunità con problemi di udito verso i test genetici sono stati recentemente
esaminati in collaborazione con l’applicazione generalizzata dello screening
uditivo neonatale. Ottantacinque percentuale di udito, e il 62% dei sordi
o con problemi di udito individui consentirebbe analisi genetica per il loro
bambino, che punta ad aumentare l’accettazione dei test genetici per la perdita
dell’udito. 58
Trattamento
La sordità è il solo difetto sensoriale che può essere trattata
con successo anche se sordità è completo. Un recente studio impianto
cocleare in bambini di 8 a 9 anni di età che hanno ricevuto i loro impianti,
prima dei cinque anni, ha dimostrato che tutti i bambini hanno beneficiato di
impianto cocleare nei settori della produzione del linguaggio, la percezione
del parlato e la lingua. C’era una differenza positiva significativa nella
performance cognitiva e la lettura nei bambini con identificati GJB2 mutazioni
che causano un insulto isolato alla coclea senza danni al nervo VIII o il
sistema uditivo centrale. Anche se la perdita di udito in altri bambini
può essere non sindromica e isolato in apparenza, le eziologie sottostanti sono
suscettibili di includere asintomatica citomegalovirus congenito (CMV) e la
meningite non diagnosticata. Così, questi bambini rischiano di affrontare
SNHL con lievi minorazioni aggiuntive a causa di effetti centrali. 59
Chirurgia impianto cocleare è stata eseguita anche in pazienti
con MELAS, ereditarietà materna diabete e sordità, sindrome di Kearns-Sayre, e
oftalmoplegia esterna progressiva cronica. Anche se una varietà di
mutazioni può causare la perdita dell’udito mitocondriale e, anche se la
gravità variabile così come progressione dopo l’esordio iniziale sono
caratteristiche, la chirurgia impianto cocleare è stato molto utile.Questo
suggerisce fortemente che i cambiamenti patologici derivanti dalle mutazioni
del mtDNA colpiscono soprattutto la coclea. 42
Conclusioni
Il campo di perdita dell’udito e della sordità ha fatto grandi
passi avanti nei settori della ricerca, screening neonatale, diagnosi
molecolare e trattamento. La perdita dell’udito è ora identificato presto
in tutti i 50 stati, e dei primi risultati di conferma della possibilità di
utilizzo più inclusivo del linguaggio e della parola. Attraverso un
maggiore indice di sospetto, cause sindromiche possono essere diagnosticati o
esclusi con una attenta valutazione, e la base molecolare della sordità può
essere determinato in modo più affidabile che mai. Nei casi di SNHL non
sindromica, GJB2 analisi della mutazione deve sempre essere
offerto, preferibilmente in combinazione graduale con GJB6 test. Eredità
e test mitocondriale devono essere considerati in ogni famiglia con più persone
affette, tranne quando la perdita di udito è stato chiaramente trasmessa
attraverso un maschio. Quando più saggi diventano disponibili, test
molecolari potrebbe diventare il primo passo verso la determinazione causale
durante il test più invasivi potrebbe essere evitato. Una volta stabilita
la causa, il trattamento come ad esempio l’impianto cocleare può migliorare
notevolmente la comunicazione e la qualità della vita di molti
pazienti. Allo stesso tempo, ogni scoperta nella biologia del sistema
uditivo ci porta un passo più vicini a trasformare il silenzio al suono.
References
-
- Petit, C, Levilliers,
J, Marlin, S, and Hardelin, J-P. Hereditary hearing loss. in: CR
Scriver, AL Beaudet, WS Sly, D Valle (Eds.) The Metabolic and
Molecular Bases of Inherited Disease. 4. ed
8. McGraw-Hill,New York; 2001: 6281–6328
- Petit, C, Levilliers,
-
- Mason, JA and
Herrmann, KR. Universal infant hearing screening by automated
auditory brainstem response measurement. Pediatrics. 1998; 101: 221–228
- Mason, JA and
-
- | CrossRef
-
- | PubMed
-
- Parving, A. The
need for universal neonatal hearing screening: some aspects of
epidemiology and identification. Acta Paediatr Suppl. 1999; 88: 69–72
- Parving, A. The
-
- | CrossRef
-
-
- | PubMed
-
-
- Centers for Disease
Control and Prevention (CDC). Infants tested for hearing loss:
United States, 1999–2001. MMWR. 2003; 52: 981–984
- Centers for Disease
-
-
- | PubMed
-
-
- Yoshinaga-Itano,
C, Sedey, AL, Coulter, DK, and Mehl, AL. Language of early- and
later-identified children with hearing loss. Pediatrics. 1998; 102: 1161–1171
- Yoshinaga-Itano,
-
- | CrossRef
-
- | PubMed
-
- Baroch, KA. Universal
newborn hearing screening: fine-tuning the process. Curr Opin Otolaryngol
Head Neck Surg. 2003; 11: 424–427
- Baroch, KA. Universal
-
- | CrossRef
-
- | PubMed
-
- Marres, HA. Congenital
abnormalities of the inner ear. in: H Ludman, T Wright, Bath
(Eds.) Diseases
of the Ear. Arnold
& Oxford University Press, ; 1998: 288–296
- Marres, HA. Congenital
-
- Friedman, TB and
Griffith, AJ. Human nonsyndromic sensorineural deafness. Annu
Rev Genomics Hum Genet. 2003; 4: 341–402
- Friedman, TB and
-
- | CrossRef
-
- | PubMed
-
- Van Camp, G, Willems,
PJ, and Smith, RJ. Nonsyndromic hearing impairment: unparalleled
heterogeneity. Am
J Hum Genet. 1997; 60: 758–764
- Van Camp, G, Willems,
-
-
- | PubMed
-
-
- ACMG. Genetics
evaluation guidelines for the etiologic diagnosis of congenital hearing
loss. Genetic Evaluation of Congenital Hearing Loss Expert Panel: ACMG
statement. Genet
Med. 2002; 4:162–171
- ACMG. Genetics
-
- | CrossRef
-
- | PubMed
-
- Hone, SW and Smith,
RJ. Medical evaluation of pediatric hearing loss: laboratory,
radiographic, and genetic testing. Otolaryngol Clin North
Am. 2002; 35: 751–764
- Hone, SW and Smith,
-
- | Abstract
-
- | PubMed
-
- Li, XC and Friedman,
RA. Nonsyndromic hereditary hearing loss. Otolaryngol
Clin North Am. 2002;35: 275–285
- Li, XC and Friedman,
-
- | Abstract
-
- | PubMed
-
- Van Laer, L, Cryns, K,
Smith, RJ, and Van Camp, G. Nonsyndromic hearing loss. Ear Hear. 2003; 24:275–288
- Van Laer, L, Cryns, K,
-
- | CrossRef
-
- | PubMed
-
- Kelsell, DP, Dunlop,
J, Stevens, HP, Lench, NJ, Liang, JN, Parry, G, Mueller, RF, and Leigh,
IM.Connexin 26 mutations in hereditary non-syndromic sensorineural
deafness. Nature. 1997; 387: 80–83
- Kelsell, DP, Dunlop,
-
- | CrossRef
-
-
- | PubMed
-
-
- Del Castillo, I,
Moreno-Pelayo, MA, Del Castillo, FJ, Brownstein, Z, Marlin, S, Adina, Q,
Cockburn, DJ, Pandya, A, Siemering, KR, Chamberlin, GP, Ballana, E, Wuyts,
W, Maciel-Guerra, AT, Alvarez, A, Villamar, M, Shohat, M, Abeliovich, D,
Dahl, HH, Estivill, X, Gasparini, P, Hutchin, T, Nance, WE, Sartorato, EL,
Smith, RJ, Van Camp, G, Avraham, KB, Petit, C, and Moreno, F. Prevalence
and evolutionary origins of the del(GJB6–D13S1830) mutation in the DFNB1
locus in hearing-impaired subjects: a multi-center study. Am J Hum Genet. 2003; 73: 1452–1458
- Del Castillo, I,
-
- | Abstract
-
- | PubMed
-
- Denoyelle,
F, Weil, D, Maw, MA, Wilcox, SA, Lench, NJ, Allen-Powell, DR, Osborn, AH,
Dahl, HH, Middleton, A, Houseman, MJ, Dode, C, Marlin, S, Boulila-ElGaied,
A, Grati, M, Ayadi, H, BenArab, S, Bitoun, P, Lina-Granade, G, Godet, J,
Mustapha, M, Loiselet, J, El-Zir, E, Aubois, A, Joannard, A, Levilliers,
J, Garabedian, E, Mueller, RF, Gardner, RJM, and Petit, C. Prelingual
deafness: high prevalence of a 30delG mutation in the connexin 26
gene. Hum Mol Genet. 1997; 6: 2173–2177
- Denoyelle,
-
- | CrossRef
-
- | PubMed
-
- Van Laer, L, Coucke,
P, Mueller, RF, Caethoven, G, Flothmann, K, Prasad, SD, Chamberlin, GP,
Houseman, M, Taylor, GR, Van de Heyning, CM, Fransen, E, Rowland, J,
Cucci, RA, Smith, RJ, and Van Camp, G. A common founder for the
35delG GJB2 gene mutation in connexin 26 hearing impairment. J Med Genet. 2001; 38: 515–518
- Van Laer, L, Coucke,
-
- | CrossRef
-
-
- | PubMed
-
-
- Green, GE, Scott, DA,
McDonald, JM, Woodworth, GG, Sheffield, VC, and Smith, RJ. Carrier
rates in the midwestern United States for GJB2 mutations causing inherited
deafness. JAMA. 1999; 281:2211–2216
- Green, GE, Scott, DA,
-
- | CrossRef
-
- | PubMed
-
- Fitzgerald, T, Duva,
S, Ostrer, H, Pass, K, Oddoux, C, Ruben, R, and Caggana, M. The
frequency of GJB2 and GJB6 mutations in the New York State newborn
population: feasibility of genetic screening for hearing defects. Clin Genet. 2004; 65: 338–342
- Fitzgerald, T, Duva,
-
- | CrossRef
-
- | PubMed
-
- Cohn, ES and Kelley,
PM. Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2),
the most common cause of childhood hearing loss. Am J Med Genet. 1999; 89: 130–136
- Cohn, ES and Kelley,
-
- | CrossRef
-
- | PubMed
-
- Yan, D, Park, HJ,
Ouyang, XM, Pandya, A, Doi, K, Erdenetungalag, R, Du, LL, Matsushiro, N,
Nance, WE, Griffith, AJ, and Liu, XZ. Evidence of a founder effect
for the 235delC mutation of GJB2 (connexin 26) in east Asians. Hum Genet. 2003; 114: 44–50
- Yan, D, Park, HJ,
-
- | CrossRef
-
- | PubMed
-
- Grifa,
A, Wagner, CA, D’Ambrosio, L, Melchionda, S, Bernardi, F, Lopez-Bigas, N,
Rabionet, R, Arbones, M, Monica, MD, Estivill, X, Zelante, L, Lang, F, and
Gasparini, P. Mutations in GJB6 cause nonsyndromic autosomal
dominant deafness at DFNA3 locus. Nat Genet. 1999; 23: 16–18
- Grifa,
-
- | CrossRef
-
- | PubMed
-
- Del
Castillo, I, Villamar, M, Moreno-Pelayo, MA, del Castillo, FJ, Alvarez, A,
Telleria, D, Menendez, I, and Moreno, F. A deletion involving the
connexin 30 gene in nonsyndromic hearing impairment. N Engl J
Med. 2002; 346: 243–249
- Del
-
- | CrossRef
-
- | PubMed
-
- Wu, BL, Kenna, M, Lip,
V, Irons, M, and Platt, O. Use of a multiplex PCR/sequencing
strategy to detect both connexin 30 (GJB6) 342 kb deletion and connexin 26
(GJB2) mutations in cases of childhood deafness. Am J Med Genet. 2003; 121A: 102–108
- Wu, BL, Kenna, M, Lip,
-
- | CrossRef
-
-
- | PubMed
-
-
- Stevenson, VA, Ito, M,
and Milunsky, JM. Connexin-30 deletion analysis in connexin-26
heterozygotes. Genet
Test. 2003; 7: 151–154
- Stevenson, VA, Ito, M,
-
- | CrossRef
-
- | PubMed
-
- Brown, CW, Levy, ML,
Flaitz, CM, Reid, BS, Manolidis, S, Hebert, AA, Bender, MM, Heilstedt, HA,
Plunkett, KS, Fang, P, Roa, BB, Chung, P, Tang, HY, Richard, G, and
Alford, RL. A novel GJB2 (connexin 26) mutation, F142L, in a
patient with unusual mucocutaneous findings and deafness. J Invest Dermatol. 2003; 121: 1221–1223
- Brown, CW, Levy, ML,
-
- | CrossRef
-
- | PubMed
-
- Lamartine,
J, Munhoz Essenfelder, G, Kibar, Z, Lanneluc, I, Callouet, E, Laoudj, D,
Lemaitre, G, Hand, C, Hayflick, SJ, Zonana, J, Antonarakis, S,
Radhakrishna, U, Kelsell, DP, Christianson, AL, Pitaval, A, Der
Kaloustian, V, Fraser, C, Blanchet-Bardon, C, Rouleau, GA, and Waksman,
G. Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat
Genet. 2000; 26: 142–144
- Lamartine,
-
- | CrossRef
-
- | PubMed
-
- Smith, FJ, Morley, SM,
and McLean, WH. A novel connexin 30 mutation in Clouston
syndrome. J Invest Dermatol. 2002; 118: 530–532
- Smith, FJ, Morley, SM,
-
- | CrossRef
-
- | PubMed
-
- Richard, G. Connexin
gene pathology. Clin
Exp Dermatol. 2003; 28: 397–409
- Richard, G. Connexin
-
- | CrossRef
-
- | PubMed
-
- Simon, AM and
Goodenough, DA. Diverse functions of vertebrate gap
junctions. Trends
Cell Biol.1998; 8: 477–483
- Simon, AM and
-
- | Abstract
-
- | PubMed
-
- Battey, JF Jr. Using
genetics to understand auditory function and improve diagnosis. Ear Hear.2003; 24: 266–269
- Battey, JF Jr. Using
-
- | CrossRef
-
- | PubMed
-
- Saez, JC, Berthoud,
VM, Branes, MC, Martinez, AD, and Beyer, EC. Plasma membrane
channels formed by connexins: their regulation and functions. Physiol Rev. 2003; 83: 1359–3400
- Saez, JC, Berthoud,
-
- | CrossRef
-
-
- | PubMed
-
-
- Goodenough, DA,
Goliger, JA, and Paul, DL. Connexins, connexons, and intercellular
communication. Annu
Rev Biochem. 1996; 65: 475–502
- Goodenough, DA,
-
- | CrossRef
-
-
- | PubMed
-
-
- Rabionet, R,
Gasparini, P, and Estivill, X. Molecular genetics of hearing
impairment due to mutations in gap junction genes encoding beta
connexins. Hum
Mutat. 2000; 16: 190–202
- Rabionet, R,
-
- | CrossRef
-
- | PubMed
-
- Teubner, B, Michel, V,
Pesch, J, Lautermann, J, Cohen-Salmon, M, Sohl, G, Jahnke, K, Winterhager,
E, Herberhold, C, Hardelin, JP, Petit, C, and Willecke, K. Connexin30
(Gjb6)-deficiency causes severe hearing impairment and lack of
endocochlear potential. Hum
Mol Genet. 2003; 12: 13–21
- Teubner, B, Michel, V,
-
- | CrossRef
-
- | PubMed
-
- Rouan, F, White, TW,
Brown, N, Taylor, AM, Lucke, TW, Paul, DL, Munro, CS, Uitto, J, Hodgins,
MB, and Richard, G. Trans-dominant inhibition of connexin-43 by
mutant connexin-26: implications for dominant connexin disorders affecting
epidermal differentiation. J Cell Sci. 2001; 114: 2105–2113
- Rouan, F, White, TW,
-
-
- | PubMed
-
-
- Marziano, NK,
Casalotti, SO, Portelli, AE, Becker, DL, and Forge, A. Mutations
in the gene for connexin 26 (GJB2) that cause hearing loss have a
dominant-negative effect on connexin 30. Hum Mol Genet. 2003; 12: 805–812
- Marziano, NK,
-
- | CrossRef
-
- | PubMed
-
- Everett, LA, Glaser,
B, Beck, JC, Idol, JR, Buchs, A, Heyman, M, Adawi, F, Hazani, E, Nassir,
E, Baxevanis, AD, Sheffield, VC, and Green, ED. Pendred syndrome
is caused by mutations in a putative sulphate transporter gene
(PDS). Nat
Genet. 1997; 17: 411–422
- Everett, LA, Glaser,
-
- | CrossRef
-
-
- | PubMed
-
-
- Li, XC, Everett, LA,
Lalwani, AK, Desmukh, D, Friedman, TB, Green, ED, and Wilcox, ER. A
mutation in PDS causes non-syndromic recessive deafness. Nat
Genet. 1998; 18: 215–217
- Li, XC, Everett, LA,
-
- | CrossRef
-
-
- | PubMed
-
-
- Reardon, W, Coffey, R,
Phelps, PD, Luxon, LM, Stephens, D, Kendall-Taylor, P, Britton, KE,
Grossman, A, and Trembath, R. Pendred syndrome: 100 years of
under-ascertainment?. Q
J Med. 1997; 90: 443–447
- Reardon, W, Coffey, R,
-
-
- | CrossRef
-
-
- Robertson,
NG, Lu, L, Heller, S, Merchant, SN, Eavey, RD, McKenna, M, Nadol, JB Jr,
Miyamoto, RT, Linthicum, FH Jr, Lubianca Neto, JF, Hudspeth, AJ, Seidman,
CE, Morton, CC, and Seidman, JG.Mutations in a novel cochlear gene
cause DFNA9, a human nonsyndromic deafness with vestibular
dysfunction. Nat Genet. 1998; 20: 299–303
- Robertson,
-
- | CrossRef
-
- | PubMed
-
- Sinnathuray, AR, Raut,
V, Awa, A, Magee, A, and Toner, JG. A review of cochlear
implantation in mitochondrial sensorineural hearing loss. Otol Neurotol. 2003; 24: 418–426
- Sinnathuray, AR, Raut,
-
- | CrossRef
-
- | PubMed
-
- Fischel-Ghodsian,
N. Mitochondrial deafness. Ear Hear. 2003; 24: 303–313
- Fischel-Ghodsian,
-
- | CrossRef
-
- | PubMed
-
- Sue, CM, Tanji, K,
Hadjigeorgiou, G, Andreu, AL, Nishino, I, Krishna, S, Bruno, C, Hirano, M,
Shanske, S, Bonilla, E, Fischel-Ghodsian, N, DiMauro, S, and Friedman,
R. Maternally inherited hearing loss in a large kindred with a
novel T7511C mutation in the mitochondrial DNA tRNA(Ser(UCN)) gene.Neurology. 1999; 52: 1905–1908
- Sue, CM, Tanji, K,
-
- | CrossRef
-
-
- | PubMed
-
-
- Bitner-Glindzicz, M,
Turnpenny, P, Hoglund, P, Kaariainen, H, Sankila, EM, van der Maarel, SM,
de Kok, YJ, Ropers, HH, Cremers, FP, Pembrey, M, and Malcolm, S. Further
mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked
deafness, DFN3. Hum
Mol Genet. 1995; 4: 1467–1469
- Bitner-Glindzicz, M,
-
- | CrossRef
-
-
- | PubMed
-
-
- Strom, TM, Hortnagel,
K, Hofmann, S, Gekeler, F, Scharfe, C, Rabl, W, Gerbitz, KD, and
Meitinger, T.Diabetes insipidus, diabetes mellitus, optic atrophy, and
deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding
for a predicted transmembrane protein. Hum Mol Genet. 1998; 7:2021–2028
- Strom, TM, Hortnagel,
-
- | CrossRef
-
-
- | PubMed
-
-
- Bespalova, IN, Van
Camp, G, Bom, SJ, Brown, DJ, Cryns, K, DeWan, AT, Erson, AE, Flothmann, K,
Kunst, HP, Kurnool, P, Sivakumaran, TA, Cremers, CW, Leal, SM, Burmeister,
M, and Lesperance, MM.Mutations in the Wolfram syndrome 1 gene (WFS1)
are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet. 2001; 10: 2501–2508
- Bespalova, IN, Van
-
- | CrossRef
-
-
- | PubMed
-
-
- Cryns, K, Sivakumaran,
TA, Van den Ouweland, JM, Pennings, RJ, Cremers, CW, Flothmann, K, Young,
TL, Smith, RJ, Lesperance, MM, and Van Camp, G. Mutational
spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing
impairment, diabetes mellitus, and psychiatric disease.Hum Mutat. 2003; 22: 275–287
- Cryns, K, Sivakumaran,
-
- | CrossRef
-
- | PubMed
-
- Guilford, P, Ben Arab,
S, Blanchard, S, Levilliers, J, Weissenbach, J, and Belkahia, A. Joint
Committee on Infant Hearing; American Academy of Audiology; American
Academy of Pediatrics; American Speech-Language-Hearing Association;
Directors of Speech and Hearing Programs in State Health and Welfare
Agencies. Year 2000 position statement: principles and guidelines for
early hearing detection and intervention programs. Pediatrics. 2000; 106: 798–817
- Guilford, P, Ben Arab,
-
- | CrossRef
-
-
- | PubMed
-
-
- Green, GE, Smith, RJ,
Bent, JP, and Cohn, ES. Genetic testing to identify deaf
newborns. JAMA.2000; 284: 1245
- Green, GE, Smith, RJ,
-
- | CrossRef
-
-
- | PubMed
-
-
- Armitage, IM, Burke,
JP, and Buffin, JT. Visual impairment in severe and profound
sensorineural deafness. Arch
Dis Child. 1995; 73: 53–56
- Armitage, IM, Burke,
-
- | CrossRef
-
-
- | PubMed
-
-
- Kenneson, A, Van
Naarden Braun, K, and Boyle, C. GJB2 (connexin 26) variants and
nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 2002; 4: 258–274
- Kenneson, A, Van
-
- | CrossRef
-
- | PubMed
-
- Wu, BL, Lindeman, N,
Lip, V, Adams, A, Amato, RS, Cox, G, Irons, M, Kenna, M, Korf, B, Raisen,
J, and Platt, O. Effectiveness of sequencing connexin 26 (GJB2) in
cases of familial or sporadic childhood deafness referred for molecular
diagnostic testing. Genet
Med. 2002; 4: 279–288
- Wu, BL, Lindeman, N,
-
- | CrossRef
-
- | PubMed
-
- Pandya, A, Arnos, KS,
Xia, XJ, Welch, KO, Blanton, SH, Friedman, TB, Garcia Sanchez, G, Liu,
MDXZ, Morell, R, and Nance, WE. Frequency and distribution of GJB2
(connexin 26) and GJB6 (connexin 30) mutations in a large North American
repository of deaf probands. Genet Med. 2003; 5: 295–303
- Pandya, A, Arnos, KS,
-
- | CrossRef
-
- | PubMed
-
- Steel, KP. A
new era in the genetics of deafness. N Engl J Med. 1998; 339: 1545–1547
- Steel, KP. A
-
- | CrossRef
-
- | PubMed
-
- Middleton, A, Hewison,
J, and Mueller, RF. Attitudes of deaf adults toward genetic
testing for hereditary deafness. Am J Hum Genet. 1998; 63: 1175–1180
- Middleton, A, Hewison,
-
- | Abstract
-
- | PubMed
-
- Middleton, A, Hewison,
J, and Mueller, R. Prenatal diagnosis for inherited deafness: what
is the potential demand?. J Genet Couns. 2001; 10: 121–131
- Middleton, A, Hewison,
-
- | CrossRef
-
-
- | PubMed
-
-
- Martinez,
A, Linden, J, Schimmenti, LA, and Palmer, CG. Attitudes
of the broader hearing, deaf, and hard-of-hearing community toward genetic
testing for deafness. Genet
Med. 2003; 5: 106–112
- Martinez,
-
- | CrossRef
-
- | PubMed
-
- Bauer, PW, Geers, AE,
Brenner, C, Moog, JS, and Smith, RJ. The effect of GJB2 allele
variants on performance after cochlear implantation. Laryngoscope. 2003; 113: 2135–2140
- Bauer, PW, Geers, AE,
-
- | CrossRef
-
- | PubMed