Proctor e ColI, in una vasta analisi dei lavori scientifici sulle sordità progressive pubblicati negli anni 1952-59, prendono in esame innumerevoli cause di sordità progressiva etra queste un certo spazio e anche dedicato alle forme ereditarie Di particolare interesse assumono queste affermazioni di van Egmond “There are two types of congenital hereditary deafness:
1) dominant hereditary, 2) recessive sporadic Dominant inner-ear deafness is rarely congenital. It develops in middle age, has a strong progression with frequent lengthy pauses, and a loss of hearing for ali frequencies
I progressi della biologia e della genetica molecolare hanno dato un contributo importante alla comprensione della fisiologia e fisiopatologia dell’orecchio interno. Questo va, e non solo per le forme profonde e congenite d’ipoacusia neurosensoriale di cui sono noti i ruoli di connessine, miosine, pendrina e usherine, ma anche per le forme progressive ad insorgenza tardiva si ricordi il ruolo dei geni COL4A nella membrana basilare nella sindrome di Alport, il gene della glomerulosclerosi Mpvl7, deficienza che conduce a notevoli alterazioni delle strutture dell’orecchio interno e il gene COCH (nei pazienti con mutazioni nel gene COCH, si ha deposizione di sostanze acidofile nell’orecchio interno che può causare la degenerazione di assoni e dendriti cocleari e vestibolari). Le ipoacusie geneticamente determinate si possono sviluppare a qualsiasi età nel corso della vita, sia come manifestazione unica del gene mutante, sia come parte di una sindrome ereditaria, oppure come risultato dell’interazione con fattori esogeni è infatti possibile che le mutazioni in alcuni geni rendano l’orecchio più sensibile a fattori ambientali che causano un danno dell’orecchio interno (ad esempio esposizione al rumore, infezioni, lesioni e farmaci ototossici), Anche se la maggior parte delle forme genetiche sono congenite, una serie di anomalie dell’udito appaiono durante o dopo il primo decennio di vita e sono progressive.
I IPOOACUSIE GENETICHE NON SINDROMICHE PROGRESSIVE
► Definizione: Per le sordità congenite, la cui epidemiologia è meglio conosciuta, si stima attualmente che tre quarti delle (1)sordità siano di origine genetica, mentre le altre cause sono ambientali. La sordità è la disabilità sensoriale più frequente e nella maggior parte dei casi l’eziologia è genetica. Questo stravolgimento nella stima della parte genetica delle sordità è dovuto al fatto che lo sviluppo delle diagnosi molecolari ha permesso di ricollegare a una causa genetica la maggioranza dei casi sporadici di sordità, in precedenza classificati come «causa ignota». i disturbi della funzione dell’orecchio interno definiti nel complesso come ipoacusia/sordità “congenita” possono insorgere in epoca prenatale, perinatale o post natale (entro i primi 6 mesi di vita) . Decine di geni sono responsabili di forme pre- o postlinguali di sordità isolata (non sindromica), e sono state descritte numerose centinaia di sindromi con sordità. A tutt’oggi sono identificati 33 geni (e quattro mutazioni mitocondriali) per le sordità non sindromiche, e più di 100 per le sordità sindromiche. È stata sviluppata una diagnosi molecolare di routine per alcuni di questi geni, in particolare per il gene della connexina 26, GJB2, gene principalmente in causa nelle sordità congenite. Una valutazione clinica e laboratoristica sistematica è particolarmente importante per orientare la strategia della diagnosi molecolare e la consulenza genetica. Le patologie genetiche possono essere classificate in base al difetto in cromosomiche (o genomiche). monogeniche o mendeliane, mitocondriali e poligeniche/multifattoriali (Fig. 1).
Le cromosomiche a loro volta possono essere classificate in anomalie di numero (aneuploidie) o di struttura. mentre le monogeniche vengono suddivise in base al tipo di trasmissione mendeliana (autosomiche dominanti e recessive, X—linked dominanti o recessive). Le malattie poligeniche riconoscono una base eziologica più complessa, con il coinvolgimento di più loci genici, che contribuiscono a dar luogo ad un fenotipo oppure a predisporre. con la concausa di fattori ambientali e/o stocastici. all’espressione di un fenotipo patologico (multifattorialità)
|
|
|
|
Fig .1 distribuzione per eziologia ed età d’insorgenza della ipoacusia MARTINI A. ed Al ”le sordità infantili” Paludetti G., 2011 ed Omega |
Tab 1 incidenza e fenotipo di alcune forme sindromiche MARTINI A. ed Al., ”le sordità infantili” Paludetti G., 2011 ed Omega |


► Incidenza: sordità congenita (>40 dB): 100-300 ogni 100.000 nascite/anno.
► Cause:
● Cause prenatali:
– Ipoacusia / sordità su base genetica non associata ad altre malformazioni o malattie ereditarie:(2) ipoacusia / sordità non sindromica (circa 70% delle ipoacusie/sordità su base ereditaria, 180% delle quali (3)autosomiche recessive, 15-20% (4)autosomiche dominanti, 2% (5)legate al cromosoma X). Circa il 50% delle ipoacusie non sindromiche è collegato a mutazioni del gene connessina 26. La trasmissione è autosomica recessiva con un grado di espressività di diversa forza. Il gene codifica le proteine delle gap junctions nella stria vascolare che sono necessarie per il trasporto degli ioni potassio ed il mantenimento del potenziale endococleare.
– Ipoacusia / sordità su base genetica associata ad altre malformazioni o malattie genetiche: (6)ipoacusia/ sordità sindromica (30% delle forme ereditane), ad es. sindrome di Alport (glomerulonefrite della membrana basale, opacamento del cristallino, ipoacusia neurosensoriale bilaterale progressiva), retinite pigmentosa e sordità congenita (sindrome di Usher), mucopolisaccaridosi (sindrome di Hurler), sindrome di Lange-Jewell (sordità congenita con disturbi della conduzione cardiaca), sindrome di Pendred (sordità congenita bilaterale, gozzo tiroideo, disturbi vestibolari, evtl. displasia dell’orecchio interno tipo Mondini), sindrome di KleinWaardenburg, albinismo, alterazioni cromosorniche (ad es trisomie). o
– Malformazioni craniofacciali (ad es. sindrome di Franceschetti-Treacher- E Collins,(Fig. 2a-b-c)
![[clip_image002%255B4%255D.jpg]](http://www.otorino-tanzariello.it/images/IPOACUSIA_PROGRESSIVA_SU_BASE_GENETICA_file/image003.jpg) Fig. 2a-b-c
Fig. 2a-b-c
– Aplasia congenita dell’orecchio interno (Scheibe-, Michel-, Mondini, ). (Fig. 3)
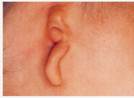 Fig. 3
Fig. 3
– Embriopatia rubeolica e altre infezioni virali intrauterine (ad es. toxoplasmosi, herpes, citomegalia).
– Lue congenita; terapia con antibiotici aminoglicosidici, alcolismo o assunzione di farmaci o droghe da parte della madre durante la gravidanza, difetti enzimatici (malattie del ricambio).
– Embriopatie farmacologiche (ad es. talidomide, clorochina).
● Cause perinatali: ad es. ipossia, asfissia, incompatibilità materno fetale (ittero nucleare), infezione da herpes durante il parto o trauma da parto con emorragia endocranica, prernaturità, malattia delle membrane ialine.
● Cause postnatali: infezioni da virus neurotropi (ad es. morbillo, parotite, rosolia), dopo vaccinazioni contro. malattie virali, meningiti batteriche, encefaliti, trauma cranio encefalico, trattamento con antibiotici aminoglicosidici.
►Modalità di trasmissione
Le sordità genetiche si possono trasmettere secondo diverse modalità.
Autosomica recessiva (AR)
Autosomica dominante (AD)
Legata al cromosoma X
Modalità di trasmissione mitocondriale
► Sintomi: sintomo principale è l’alterazione o l’assenza completa di sviluppo verbale. La compromissione uditiva porta quasi sempre ad un ritardo psichico poiché il pensiero si completa in concetti astratti per la mediazione dei quali è necessaria la comprensione del linguaggio (a mezzo della funzione uditiva). La compromissione della capacità comunicativa può anche comportare una compromissione del rapporto mamma / bambino con evidenti conseguenze psicologiche. Eventuali ulteriori sintomi sono presenti in dipendenza della causa della sordità (ad es. in caso di sindromi).
► Complicanze: il riconoscimento tardivo o assente dell’ipoacusia / sordità impedisce la maturazione delle vie acustiche centrali che si completa nei primi 12 mesi di vita.
Diagnostica
► Indispensabile:
● Osservazione accurata del neonato: assenza di reazione agli stimoli acustici. Mimica e gestualità esagerate in alternanza con fasi “autistiche”.
● Anamnesi familiare differenziata: inchiesta su eventuale presenza di sordità familiare.
● Esame obiettivo ORL: ricerca di malformazioni esterne.
● Indagini precoci di audiometria infantile (screening uditivo) con impiego di metodiche di ricerca obiettiva (emissioni otoacustiche, audiometria del tronco encefalico, potenziali evocati). Queste indagini sono possibili già sino dai primi giorni dopo la nascita. Esse sono indispensabili nei bambini
a rischio (per es. prematuri, bambini ricoverati in terapia intensiva o con storia familiare positiva).
– Otomicroscopia.
– Esame della funzionalità uditiva: in dipendenza dall’età d)emissioni otoacustiche, audiometria infantile, timpanogramma, misurazione dello riflesso dello stapedio, tecniche obiettive .
– a)L’esame oftalmologico esteso al fondo dell’occhio sistematico è indispensabile
– b)La ricerca di ematuria e l’elettrocardiogramma (ECG)
– c)TAC: tomografia computerizzata delle rocche indispensabile
– RM per evidenziare orecchio interno, nervi acustici e vie uditive centrali.
► Utile in casi particolari:
● Audiogramma possibile a partire dal terzo anno di vita circa.
● Consulto interdisciplinare: pediatra, neuropediatra, genetista (analisi cromosomica).
Diagnosi differenziale
► Neuropatia uditiva — Causa: presumibile difetto congenito o acquisito mono o bilaterale della catena sinaptica tra cellule acustiche interne e 10 neurone o nell’ambito del 1° neurone (ganglio spirale). Otoernissioni acustiche e potenziali microfonici cocleari presenti, riflesso acustico ipsilaterale e potenziali del tronco presenti. La RM mostra un reperto normale.
► Ipoacusia/sordità centrale — Causa: danni della via acustica a partire dal 1 neurone (VIII) fino ai centri più alti.
► Disturbo della percezione uditiva Causa: complessa alterazione dell’elaborazione e del riconoscimento dei suoni.
► Ritardo mentale, ritardo di sviluppo generalizzato.
TERAPIA
► Terapia medica:
● La diagnosi precoce è essenziale!
● Protesizzazione precoce binaurale possibile con apparecchi acustici retro- auricolari già entro le prime settimane di vita.
● Nel caso di assenza del padiglione auricolare e/o atresia del condotto uditivo esterno, precoce applicazione di apparecchio acustico per via ossea con archetto e precoce “allenamento acustico’ a partire dai primi mesi di vita (!).
● Allenamento linguistico precoce dal secondo anno di vita (servizio pedoaudiologico).
● Controlli regolari da parte di un pedoaudiologo (applicazione e controllo di apparecchi acustici), neuropediatra ed eventuale neuropsicologo infantile.
► Indicazione all’intervento chirurgico, principi dell’intervento:
● In caso di malformazioni dell’orecchio esterno eventuale apparecchio acustico ancorato all’osso/epitesi .
● In caso di grave perdita uditiva, intervento per impianto cocleare precoce (dai 1 anno di vita) , quando gli apparecchi acustici convenzionali non sono utilizzabili.
► Ambulatoriale/con ricovero: una diagnosi esatta dal punto di vista otorinolaringoiatrico e pedoaudiologico è possibile sia ambulatoriamente che con ricovero (insieme alla madre).
Prognosi
► Buona in caso di protesizzazione acustica precoce (pag. 204), particolarmente se associata a trattamento audiologico intensivo e a collaborazione da parte dei genitori, in assenza di altre lesioni del sistema nervoso centrale. Bisogna mirare ad una completa integrazione sociale (anche a mezzo di un impianto cocleare).
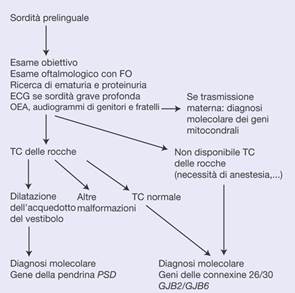
Algoritmo decisionale. Esempio di strategia diagnostica molecolare in caso di sordità prelinguale. FO: fondo dell’occhio; TC: tomografia computerizzata; ECG: elettrocardiogramma; OEA: otoemissioni acustiche.
|
Tabella 1 – Sordità autosomiche dominanti DFNA |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Esami strumentali e di laboratorio
a)L’esame oftalmologico esteso al fondo dell’occhio sistematico è indispensabile. Esso è innanzitutto utile alla diagnosi eziologica poiché più di 50 sindromi associano sordità e patologie visiva. Contribuisce inoltre allo screening di una riduzione dell’acuità visiva pregiudizievole per la lettura labiale o la comunicazione gestuale. Nella casistica di Armitage, [86] nei bambini affetti di sordità gravi e profonde, sommando insieme tutte le eziologie, l’esame oftalmologico con fondo dell’occhio è anormale nel 45,8% dei casi. In caso di ritardo della deambulazione associato a una sordità profonda nel bambino piccolo questa valutazione di base è insufficiente e deve essere completata con un elettroretinogramma, al fine di individuare precocemente i segni di retinite della sindrome di Usher e di adattare la gestione (vedi sopra) del paziente.
b)La ricerca di ematuria e l’elettrocardiogramma (ECG) sono esami semplici che possono permettere una gestione precoce delle due sindromi in cui la patologia associata può porre a rischio la prognosi quoad vitam, la sindrome di Alport e la sindrome di Jerwell e Lange-Nielsen (vedi sopra). Per la ricerca di ematuria è sufficiente uno stick urinario, e l’ECG è utile solo per le sordità congenite gravi o profonde.
c)La diagnostica per immagini della rocca sembra indispensabile. La prevalenza delle malformazioni dell’orecchio interno nei soggetti sordi è variabile in letteratura, il che si spiega con l’eterogeneità delle casistiche che raggruppano in modo differente dei bambini, degli adulti e delle sordità di eziologia diversa. Nella nostra esperienza un terzo dei bambini con una sordità familiare aveva una TC dell’orecchio interno anomala. [Denoyelle F.87] Questa percentuale sembra oggi più vicina al 20% nella coorte raggruppando ora circa 1 000 pazienti. La messa in evidenza di queste malformazioni è essenziale in quanto contribuisce ad affermare il carattere congenito della sordità nelle forme di comparsa secondaria e a orientare verso una diagnosi specifica, come la sindrome di sordità mista legata al cromosoma X con geyser-labirinto. [Talbot J.M., 88] La scoperta peraltro di malformazioni importanti dell’orecchio interno deve indurre a una vaccinazione antipneumococcica e anti- Haemophilus influenzae per prevenire il rischio di meningite. Per tutte queste ragioni, noi raccomandiamo la diagnostica per immagini della rocca in tutte le ipoacusie neurosensoriali del bambino. La TC è in genere l’esame più accessibile, poiché una RM di qualità della rocca è più difficile da ottenere. Questo esame deve essere richiesto a un radiologo formato a tale esercizio. Esso viene effettuato senza iniezione di mezzo di contrasto, in sezioni millimetriche assiali e, se possibile, coronali. L’anestesia generale non è giustificata per la sua realizzazione, poiché l’esame può attendere in genere l’età di 3-4 anni. L’indagine genetica è quindi talora realizzata senza disporre della diagnostica per immagini.
d)Tra gli altri esami utili conviene ricordare le otoemissioni acustiche, la cui realizzazione non deve essere riservata allo screening. In effetti l’esistenza di sordità per mutazione del gene dell’otoferlina (OTOF, DFNB9, vedi sopra), sordità in cui le otoemissioni acustiche sono spesso inizialmente presenti, benché la sordità sia da grave a profonda, richiede di realizzare questo esame in tutti i sordi (se non è stato fatto nel quadro della diagnosi) per isolare queste forme e richiedere una diagnosi molecolare del gene OTOF .
Infine, il ruolo dell’ORL è anche quello di praticare degli audiogrammi sistematici ai genitori e ai fratelli.
Al termine di tale valutazione, deve essere proposta alla famiglia una visita di consulenza genetica, che utilizzerà il complesso degli esami richiesti e in particolare degli esami audiometrici familiari per guidare le domande del genetista (Figura 11 ).
IPOACUSIE NON-SINDROMICHE PROGRESSIVE
Rappresentano il 70% delle ipoacusie genetiche e, tra queste, le forme autosomiche recessive sono le più frequenti e si manifestano in genere con ipoacusia alla nascita. Nelle forme dominanti, L’ipoacusia è il più delle volte progressiva o a comparsa ritardata, perfino in età adulta. La maggior parte delle ipoacusie neurosensoriali post-linguali non sindromiche sono autosomiche dominanti Anche se in letteratura sono riportati molti casi di famiglie con mutazione autosomica dominante (AD) che hanno manifestato forme d’ipoacusia progressiva non esistono dati epidemiologici sulla prevalenza delle ipoacusie genetiche nella popolazione adulta. La DFNA1 è la tipica forma (ipoacusia di ‘Monge’) in cui sono coinvolte le basse frequenze durante i primi anni, si ha la progressione della compromissione dell’udito che comporta il coinvolgimento delle alte frequenze negli anni successivi. Individui più anziani con questo genotipo mostrano una perdita uditiva che coinvolge tutte le frequenze. Questo vale anche per alcuni altri disordini non sindromici dell’udito che all’inizio possono avere differenti profili audiometrici (ad esempio, che coinvolgono medie o alte frequenze) che diventano sovrapponibili in età adulta, ad esempio grave ipoacusia pantonale con possibile caduta sulle alte frequenze Si definisce “ipoacusia progressiva” una perdita di 15 dBHL nel PTA in un periodo di 10 anni (European Work Group on Genetic Hearing lmpairment, 2000).
Studi recenti hanno identificato tre possibili geni responsabili di ipoacusia neurosensoriale progressiva a trasmissione autosomico-recessiva LOXHD1 (lipoxygenase homology domains 1) PJVK (pejvakin) e MYO3A (miosina 3A), espressi nelle cellule ciliate dell’orecchio interno ed in particolare PJVK (che codifica per una proteina citoplasmatica) è espresso a livello del ganglio spirale Questi tre geni sono indispensabili per la normale fisiologia delle cellule ciliate dell’orecchio interno Nei modelli animali, mutazioni a carico del gene LOXHD1 alterano la normale funzionalità delle cellule ciliate; studi condotti sul DNA di famiglie affette da ipoacusia progressiva hanno permesso di individuare una mutazione responsabile di DFNB77, a trasmissione autosomica recessiva, e il gene umano corrispondente è stato localizzato sul braccio lungo del cromosoma 18 (Grillet et al .., 2009). Altre forme progressive autosomico-recessive sono attribuite a mutazioni dei geni PVJK e MYO3A: rispettivamente DFNB59, caratterizzata da neuropatia (Delmaghani et al.., 2006) e DN FB3O..
Altri geni le cui mutazioni possono determinare ipoacusia neurosensoriale progressiva sono MYH9 (Myosin, heavy chain 9) e WFS1 (sindrome di Wolfram o DIDMOAD) In particolare mutazioni a carico del gene MYH9, che codifica per la catena pesante della miosina non-muscolare IIA, sono responsabili di fenotipi riconducibili alla sindrome di Epstein, di Fechtner, di Sebastian e all’anomalia di May-Hegglin. Tale classificazione è da ritenersi tuttavia superata perchè non in grado di descrivere la complessità dei quadri fenotipici conseguenti alle mutazioni del gene A quadri clinici così ampi e variabili è stato dato il nome di disordini MYH9-correlati (o malattia MYH-9 correlata), caratterizzati da grandi piastrine e in bassa concentrazione, glomerulonefrite e ipoacusia neurosensoriale progressiva per le alte frequenze che insorge in età giovane adulta, Tutti i pazienti presentano alla nascita piastrinopenia, ma alcuni di loro sono destinati a sviluppare negli anni successivi glomerulonefrite cronica e/o deficit uditivo neurosensoriale e/o cataratta presenile Il pattern clinico è ereditato in maniera autosomica dominante, A trasmissione autosomica recessiva è invece la sindrome di Wolfram (o DIDMOAD, nota anche come DFNA 6/14/38), dovuta a mutazioni a carico del gene WFS1, caratterizzata da una perdita progressiva sulle basse frequenze, dopo un iniziale decadimento sui toni acuti, disordini neurodegenerativi progressivi, diabete mellito e atrofia ottica prima dei 15 anni, atassia, neuropatia periferica e altre alterazioni neurologiche.
1.10 (1)Le sordità genetiche sono, nella maggior parte dei casi, malattie monogeniche: la lesione di un singolo gene è chiamata in causa in ciascuna forma di sordità, e la deficienza uditiva è dovuta nella grande maggioranza dei casi a una lesione cocleare. Le diverse forme di sordità non sindromiche sono in genere state caratterizzate da un’analisi di legame genetico in grandi famiglie, permettendo di definire un locus, regione del genoma contiene il gene interessato. Il gene è spesso stato identificato in un secondo tempo e, in qualche caso, un locus ha dimostrato di contenere due geni della sordità. È stata fissata una codifica internazionale per denominare ogni locus della sordità non sindromica. Per convenzione e conseguentemente alla scoperta dei loci, il codice inizia con DFNA (per deafness, autosomica dominante) o con DFNB (per deafness, autosomica recessiva), o con DFN (per deafness, legata al cromosoma X). Si attribuisce in seguito un numero per ordine di scoperta: DNFB da 1 a 40, DFNA da 1 a 48 (Tabella 1, Tabella 2, Tabella 3 ).
Tabella 2 – Sordità autosomiche recessive DFNB
|
Locus |
Gene |
Localizzazione cromosomica |
Età di comparsa |
Modalità evolutiva |
|
DFNB1 |
GJB2 (in precedenza CX26 |
13q11/13q12 |
Prelinguale |
Stabile Frequenze acute |
|
GJB6 (in precedenza CX30 |
||||
|
DFNB2 |
MYO7A |
13q12 |
Pre- o postlinguale |
Profonda stabile |
|
DFNB3 |
MYO15 |
17p11.2 |
Prelinguale |
Stabile |
|
DFNB4 |
SLC26A4 (in precedenza PDS |
7q31 |
Pre- o postlinguale |
Varia |
|
DFNB5 |
|
14q12 |
Prelinguale |
|
|
DFNB6 |
TMIE |
3p14-p21 |
|
Stabile |
|
DFNB7/11 |
TMC1 |
9q13-q21 (sordità lieve o media tra 0 e 10 anni che diviene profonda predominando sugli acuti tra 10 e 20 anni) (India – famiglia di Beduini) |
Prelinguale |
Progressiva Frequenze acute |
|
DFNB8/10 |
TMPRSS3 |
21q22.3 |
Postlinguale |
Rapidamente progressiva |
|
DFNB9 |
OTOF |
2p22-p23 |
prelinguale |
Stabile |
|
Falsa neuropatia uditiva |
||||
|
DFNB1 2 |
CDH23 |
10q21-q22 |
Prelinguale |
Stabile |
|
Tutte le frequenze |
||||
|
DFNB1 3 |
|
7q34-36 |
Prelinguale |
Progressiva |
|
DFNB14 |
|
7q31 |
Prelinguale |
Stabile |
|
DFNB1 5 |
|
3q21-q25 – 19p13 |
Prelinguale |
Stabile |
|
DFNB1 6 |
STRC |
15q15 |
1 o decennio |
Stabile Predominanza sulle frequenze acute |
|
DFNB1 7 |
|
7q31 |
|
Profonda stabile |
|
DFNB1 8 |
USH1C |
11p15.1 |
|
|
|
DFNB20 |
|
11q25-qter |
Prelinguale |
|
|
DFNB21 |
TECTA |
11q22-q24 |
Prelinguale |
Stabile |
|
Curva a U |
||||
|
DFNB22 |
OTOA |
16p12.2 |
Prelinguale |
|
|
DFNB23 |
PCDH15 |
10p11.2-q21 |
|
|
|
DFNB26 |
|
4q31 |
|
|
|
DFNB27 |
|
2q23-q31 |
Prelinguale |
|
|
DFNB28 |
|
22q13 |
Prelinguale |
|
|
DFNB29 |
CLDN14 |
21q22.3 |
Prelinguale |
|
|
DFNB30 |
MYO3A |
10p11.1 |
2 o decennio |
Progressiva Inizialmente frequenze acute |
|
DFNB31 |
WHRN |
9q32-q34 |
Prelinguale |
Profonda |
|
Stabile |
||||
|
DFNB32 |
|
1p13.3-22.1 |
Prelinguale |
Profonda |
|
Stabile |
||||
|
DFNB33 |
|
9q34.3 |
1 o decennio |
|
|
DFNB35 |
|
14q24.1-24.3 |
Prelinguale |
Stabile |
|
Tutte le frequenze |
||||
|
DFNB36 |
ESPN |
1p36.3 |
Prelinguale |
|
|
DFNB37 |
MYO6 |
6q13 |
Prelinguale |
|
|
DFNB38 |
|
6q26-q27 |
Prelinguale |
Tutte le frequenze |
|
DFNB39 |
|
7q11.22-q21.12 |
Prelinguale |
Stabile |
|
DFNB4 0 |
|
22q |
Prelinguale |
Tutte le frequenze |
|
Senza nome del locus |
GJA1 (in precedenza CX43 ) coinvolgimento in una sordità non sindromica attualmente smentita |
|
|
|
|
Senza nome del locus |
PRES |
|
|
|
Il rapporto tra geni, fenotipo clinico e modalità di trasmissione della sordità è complesso: a seconda del tipo di mutazione, uno stesso gene può essere responsabile di una sordità autosomica recessiva (AR) e/o autosomica dominante (AD) e/o di una sordità sindromica.
Rientrano in questo caso i geni:
• della connexina 26 GJB2 (DFNB1, DFNA3, sindrome di Vohwinkel, keratosis ichthyosis deafness (KID) syndrome;
• della connexina 30 GJB6 (stesso locus del gene della connexina 26, DFNB1, DFNA3, sindrome di Clouston);
• della connexina 31 GJB3 (locus DFNB non attribuito, DFNA2);
• della miosina VIIa MYO7A (DFNB2, DFNA11, sindrome di Usher Ib);
• della pendrina PDS (DFNB4, sindrome di Pendred);
• dell’ -tectorina TECTA (DFNA8/12, DFNB21);
• del collagene 11 2 COL11A2 (DFNA13, sindrome di Stickler tipo 3, sindrome di Weissenbacher-Zweymuller);
• della wolframina WFS1 (DFNA6/ 14 e sindrome di Wolfram tipo 1);
• della miosina VI MYO6 (DFNB37, DFNA22);
• della miosina IX MYO9 (DFNA17, sindrome di May-Hegglin e sindrome di Fechtner);
• USH1C (DFNB18 e sindrome di Usher Ic);
• CDH23 (DFNB12 e sindrome di Usher Id);
• TMC1 (DFNB7/11 et DFNA36).
Le sordità genetiche si possono trasmettere secondo diverse modalità.
1.10.1.1 3)Autosomica recessiva (AR)
In questa modalità di trasmissione i due alleli del gene devono essere muti perché il soggetto sia sordo. I genitori portatori di una copia anomala (allele) del gene in causa (portatori eterozigoti) sono normoudenti e, statisticamente, un quarto dei bambini (maschio o femmina) saranno sordi, portatori di mutazioni sui due alleli del gene (omozigoti). La modalità di trasmissione autosomica recessiva è favorita dalla consanguineità. Si stima che circa tre quarti delle sordità non sindromiche si trasmettano in modalità AR e che tale modalità sia la seconda in frequenza nelle sordità sindromiche.
1.10.1.1 4)Autosomica dominante (AD)
In questa modalità di trasmissione, le persone aventi un singolo allele mutato (eterozigoti) sono sorde. La mutazione genetica dell’allele interessato può, per esempio, produrre una proteina anormale, che impedisce la funzione della proteina normale prodotta dall’allele sano.
Nelle famiglie affette da sordità autosomica dominante, uno dei genitori è sordo e porta su un singolo allele del gene la mutazione patogena, che trasmetterà a metà dei suoi figli, che saranno allora sordi. L’espressività è molto spesso variabile in questa modalità di trasmissione: più soggetti colpiti nella famiglia possono presentare delle sordità di gravità molto differente. Quando la sordità è sindromica i segni associati alla sindrome possono essere assenti o lievi in alcuni sordi della famiglia, presso la quale la sordità sembra allora isolata, e alcuni membri portatori dell’alterazione genetica possono essere udenti.
1.10.1.2 5)Legata al cromosoma X
Il gene in causa è situato sul cromosoma X. Nei maschi che posseggono solo un cromosoma X la malattia si esprime e pertanto essi sono colpiti da sordità. Essi trasmetteranno l’X portatore della mutazione genetica alle loro figlie udenti. Nelle donne l’X portatore della mutazione genetica è in generale «compensato» dal secondo X normale (sordità cosiddetta recessiva legata all’X). Raramente, come nella sindrome di Alport, la sordità è definita dominante legata al cromosoma X, e le femmine non sono allora solo in condizione di trasmettere la sordità, ma sono anche colpite, in modo meno grave dei maschi.
1.10.1.3 6)Modalità di trasmissione mitocondriale
Il genoma mitocondriale è un frammento di acido desossiribonucleico (DNA) situato fuori dal nucleo della cellula, nel mitocondrio. Esso è trasmesso dalla madre. Quando un gene della sordità è situato sul DNA mitocondriale, l’albero genealogico è caratteristico perché uomini e donne possono essere sordi, ma solamente le donne potranno trasmettere la sordità ai loro figli che sono in teoria tutti sordi nel nucleo familiare (vedi paragrafo «Sordità mitocondriali»).
Noi esamineremo le forme più frequenti.
Geni della connexina
Le connexine sono piccole proteine transmembranarie che si associano in complessi (connessoni) con funzione di canali di giunzione tra le cellule adiacenti per il
trasporto di ioni implicati in particolare nei meccanismi di eccitazione delle cellule sensoi-jali cocleari. Attualmente nei mammiferi sono note cinque connexine diverse.
Quattro geni delle connexine, espressi nell’orecchio interno, possono essere implicati in ipoacusie isolate o sindromiche:
• GJB2(connexina26.inl3qll.q12)
• GJB6 (connexina 3Oinl3qll.q12)
• GJB3 (connexina 32. in Xql3.l)
• G.JB i (connexina 31. in lp34).
GJB2 è responsabile della forma principale (50% dei casi) di ipoacusia genetica, DFNB 1: più recentemente è stato dimostrato che una ipoacusia DFNB 1 può essere dovuta anche all’associazione di una delezione di GJB6. situato nello stesso locus del GJB2. con una mutazione di GJB2.
Si riteneva inizialmente che le connexine 26 e 30 fossero coinvolte nel ricircolo del potassio nell’endolinfa e che l’ipoacusia potesse essere dovuta a un’alterazione della concentrazione di K endolinfatico. La patogenesi è meglio conosciuta grazie a modelli animali di delezione parziale o totale dei geni GJB2 e GJB6 che dimostrano che l’orecchio interno si sviluppa normalmente e che la morte cellulare sopraggiunge verso il l4 giorno postnatale nel topo. Inizialmente sono interessate le cellule di sostegno e poi le cellule ciliate. La concentrazione di potassio nell’endolinfarimane normìle tino a questa data. facendo avanzare l’ipotesi di una tossicità scatenata dalla risposta delle cellule ciliate interne alla stimolazione sonora. per esempio a causa di un mancato ricircolo del glutammato ,
Gene della connexina 26 (CX26 o GJB2)
La mutazione di GJB2 è responsabile sia della forma più comune di ipoacusia del bambino, quella autosomica recessiva DFNB 1, sia di una forma rara di ipoacusia autosomica dominante, la DFNA35. Altre mutazioni di questo gene rendono conto anche di tre sindromi rare che associano ipoacusia e interessamento cutaneo, la sindrome di Vohwinkel, l’ipoacusia con cheratosi pahnoplantare e la sindrome Keratosis Ichthyosis Deafness (KID)6 7.
Nella ipoacusia DFNB i, i soggetti sordi portano delle mutazioni sui due alleli del gene, mentre i soggetti eterozigoti sono normoudenti. L’ipoacusia è congenita e stabile nella maggioranza dei casi (un’evolutività è osservata in meno del 20% deicasi, in genere lieve) ed è più spesso profonda e pantonale8.
La TC delle rocche petrose e le prove vestibolari caloriche sono normali.
Le mutazioni individuate sono allo stato oltre 60 ma la mutazione che predomina nei paesi occidentali è la 35delG, dovuta alla delezione di una guanina in posizione 35 nella parte codificante del gene e che determina la formazione di una proteina tronca. Questa mutazione rappresenta il 55 66% degli alleli mutati in Italia, il 46% negli Stati Uniti9. I portatori eterozigoti (normoudenti) di questa mutazione sono molto frequenti nella popolazione generale. tra il 2,5 e il 4% della popolazione in Italia, il 2% negli Stati Uniti10 Il
Le mutazioni “inattivanti” che portano auna proteina tronca sono associate ad ipoacusia più grave delle mutazioni “non inattivanti’5. In particolare la mutazione frequente L9OP associata a 35 delG sull’altro allele (genotipo L9OP/35delG) è più comunemente associata ad una ipoacusia medio-lieve rispetto al genotipo 35delG!35delG.
Un’altra mutazione frequente. M34T. non risulta patogena nella maggiorparte dei casi: sono stati segnalati molti soggetti 35delG/M34T normoudenti’2.
Nel 5% dei casi si riscontra inoltre una delezione del gene della connexina 30, situato nell’intervallo del locus DFNB 1, in associazione con una mutazione del GJB2 sull’altro allele,
Le mutazioni del gene della connexina 26 sono coinvolte anche nella forma di ipoacusia autosomica dominante DFNA3. Chiaramente patogene sono le mutazioni W44C e C202F’.
Le caratteristiche dell’ipoacusia sono molto diverse a seconda delle mutazioni: peresempio. per W44C l’ipoacusia è prelinguale, da media a profonda e colpisce tutte le frequenze. mentre per C202F compare tra lO e 20 anni. colpisce inizialmente le altefrequenze ed evolve lentamente per divenire media-lieve a 50 anni.
Altri geni del/e connexine
Altri geni delle connexine possono essere responsabili di una iloacusia isolata. autosomica i ecessiva e/o dominante:
GJB6 (coliliexina 30)
• GJB3 (connexina 31).
Il gene GJB6 (connexina 30) è particolarmente importante: molto simile al gene della connexina 26, una delezione di un singolo allele di questo gene (GJB6- Dl3Sl83Odel) è frequentemente riscontrata in associazione con una mutazione eterozigote del gene della connexina 26 in soggetti sordicongeniti’4.
Sono invece implicati in ipoacusie sindromiche:
• GJB i (connexina 32), responsabile di una forma della sindrome di Charcot-MarieTooth’ 5
• GJA1 (connexina 43), coinvolto nella displasia oculo-dento-digitale in cui si riscontra una ipoacusia trasmissiva16,
La diagnosi molecolare di mutazioni del GJB2!GJB6 è la prima ad essere proposta al termine della valutazione eziologica in presenza di una ipoacusia non sindromica prelinguale se la diagnostica per immagini della rocca petrosa è normale o se non ha potuto essere realizzata a causa dellagiovane età del paziente.
Nell’adulto bisogna chiedere queste indagini molecolari quando l’ipoacusia risale all’infanzia.
Gene PDS DFNB4
Il gene della sindrome di Pendred, PDS, può essere responsabile della forma di sordità DFNB4, che ricorda in ogni punto quella della sindrome (prelinguale, progressiva o fluttuante, malformazione dell’orecchio interno), ma che resta isolata, cioè senza interessamento tiroideo. Nel 1998 e 1999 due studi hanno dimostrato il coinvolgimento di mutazioni del PDS nella forma di sordità DFNB4, in soggetti che presentano una sordità congenita profonda neurosensoriale senza gozzo né ipotiroidismo, ma con una malformazione dell’orecchio interno alla TC delle rocche (dilatazione bilaterale dell’acquedotto del vestibolo [DAV], che contiene il sacco endolinfatico, in cui il criterio è rappresentato da un acquedotto più largo di 2 mm nella parte mediana). [11, 12] Il test al perclorato era normale nei soggetti testati.
Campbell et al. [62] hanno mostrato che PDS era molto frequentemente coinvolto in alcune forme familiari di sordità con malformazioni dell’orecchio interno: mutazioni di questo gene sono messe in evidenza in quattro famiglie su cinque con malformazione di Mondini e cinque famiglie su sei con DAV. In una casistica francese molto recente le mutazioni del PDS sono riscontrate nel 42% dei pazienti con sordità e malformazione dell’orecchio interno (in quasi la metà dei casi, si è potuta identificare solo una mutazione), ed era sempre presente una DAV nei pazienti con mutazione doppia. [63]
Il riconoscimento di sordità dovute alla PDS è importante per la consulenza genetica ma anche prognostica, poiché prevedere le fluttuazioni e l’evolutività può aiutare l’orientamento educativo e far prendere delle precauzioni nei confronti dei microtraumi (alcuni sport violenti, barotraumi) in questi bambini. Il trattamento degli episodi di aggravamento della sordità non è ben codificato e si basa spesso su un trattamento con vasodilatatori/corticosteroidi come si usa nella sordità improvvisa, benché questo non sia validato, ma bisogna sicuramente evitare i ricoveri iterativi con le loro ripercussioni psicologiche e scolastiche per questi bambini il cui stato può fluttuare in tempi molto ravvicinati.
Il gene della sindrome di Pendred, PDS, può essere responsabile della ipoacusia DFNB4. che ricorda quella della sindrome (prelinguale. progressiva o fluttuante. malforniazione dell’orecchio interno con dilatazione dell’acquedotto del vestibolo che supera i 2 mm nella parte mediana) (Fig. 1), ma che resta isolata, cioè senza interessamento tiroideo2425
Il riconoscimento di ipoacusia dovuta alla PDS è importante poiché prevedere le fluttuazioni e l’evolutività può far prendere delle precauzioni nei confronti dei microtraumi (alcuni sport violenti, barotraumi) in questi bambini. Il trattamento degli episodi di aggravamento della sordità si basa spesso su vasodilatatori/corticosteroidi come si usa nella ipoacusia improvvisa, benché questo non sia validato.
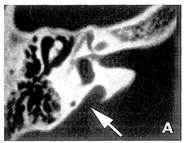
Fig. 1 — Dilatazione dell’acquedotto del vestibolo
Gene dell’otoferlina (OTOF)
Le mutazioni del gene dell’otoferlina, OTOF, sono responsabili della forma di sordità recessiva DFNB9. [64] L’otoferlina sarebbe implicata nel traffico vescicolare presinaptico delle cellule ciliate interne. [65] La sordità è grave o profonda, prelinguale, e una mutazione di questo gene, Q829X, sembra particolarmente frequente in Spagna. [Migliosi V., et al 2002]
La particolarità di questa forma di sordità è che essa può presentarsi come una neuropatia auditiva, con otoemissioni acustiche inizialmente conservate. Starr et al 1996 hanno per primi utilizzato il termine di neuropatia uditiva per designare nell’adulto delle sordità, spesso bilaterali, associate a un’alterazione dei potenziali evocati uditivi (PEU) contrastante con la presenza di otoemissioni acustiche (OEA) normali. Queste forme sono oggi meglio conosciute nell’adulto e nel bambino e giustificano la valutazione delle otoemissioni per individualizzarle nel corso della gestione di una sordità di percezione.
La frequenza di queste sordità è molto elevata (1%) nei neonati ricoverati in terapia intensiva, il che giustifica uno screening delle sordità con i potenziali evocati piuttosto che con OEA in questa popolazione. Le mutazioni dell’otoferlina sono probabilmente rare tra i bambini con una diagnosi di «neuropatia auditiva»: Madden et al 2002 tra 22 casi pediatrici, descrivevano 15 casi (68%) con patologie neonatali intricate (iperbilirubinemia: 11/15, prematurità: 10/15, farmaci ototossici: 9/15, ventilazione assistita: 8/15) e e sei casi erano rappresentati da coppie di due fratelli sordi che evocavano una causa genetica autosomica recessiva, non essendo stato ricercato il gene della DFNB9, OTOF .
Il termine neuropatia uditiva sembra assolutamente inappropriato per la sordità dovuta al gene OTOF, sordità di origine cocleare per la quale i risultati di impianto cocleare sembrano del tutto paragonabili ai risultati abituali dell’impianto. [Loundon et al., 2005]
Lo studio morfologico dell’orecchio interno e del nervo uditivo con RM nei pazienti che presentano un quadro clinico di neuropatia ha evidenziato nel 18% dei casi la presenza di un’anomalia anche anatomica del nervo acustico che appare ipoplasico, mono- o bilateralmente [Loundon et al., 2005]. E’ fondamentale chiarire la presenza di una riduzione del calibro del nervo o addirittura la sua assenza prima di procedere a intervento di impianto cocleare che nel primo caso ha opportunità di successo mentre nel secondo caso è controindicato,
Gene KCNQ4
Questo gene è espresso soprattutto nelle cellule ciliate esterne e codifica per un canale del potassio voltaggio-dipendente. Esso è coinvolto in una forma di ipoacusia progressiva, DFNA2. che interessa inizialmente le frequenze acute. L’età di esordio è variabile tra 1 e 30 anni. l’ipoacusia progredisce di circa 1 dB pe ranno. e gli acufeni sono frequenti.[[Coucke P.J., et al 1999; Roux et al., 2006 89]
Gene COH
Questo gene è espresso in grande quantità nella coclea e codifica per una proteina della matrice extracellulare cocleare, la coclina. Dal punto di vista istologico si ritrovano, in questa forma di sordità, dei depositi acidofili cocleari molto caratteristici.
L’ipoacusia dovuta alla lesione del COCH, DFNA9, ha inizio sulle frequenze acute nell’adolescenza o nell’adulto e progredisce rapidamente (3-4 dB per anno) per interessare tutte le frequenze [Verhagen et al 2001]. L’ipoacusia diventa grave-profonda in genere dopo i 60 anni.
Alcuni pazienti hanno episodi di vertigini, sensazione di “pienezza auricolare” ed acufeni che possono evocare una malattia di Ménière. anche se la forma della curva è diversa[Robertson et al 1997]..
Gene della wolframina (WFSJ)
Questo gene è stato identificato nel 2001 come responsabile di una sordità sindromica, la sindrome di Wolfram, nella quale la sordità è associata a diabete mellito e insipido e a sintomi neurologici e oculari. [Khanim et al.,2001] Due equipe hanno dimostrato che WFS1 era anche in causa in una forma non sindromica autosomica dominante, DFNA38, caratterizzata da una sordità progressiva, che inizia tra 5 e 15 anni e colpisce di preferenza le frequenze gravi. [Bespalova et al.,2005; Young et al.,2001] La sordità colpisce in seguito tutte le frequenze e diventa media/grave verso i 40 anni. I loci DFNA6, DFNA14 e DFNA38 si sovrappongono e corrispondono tutti al gene WFS1. Young et al. hanno messo in evidenza mutazioni in due famiglie statunitensi e due famiglie olandesi in cui la sordità era legata al DFNA38, ma anche in due famiglie testate senza precedente localizzazione, con sordità dominante che interessava le basse frequenze. [Young et al.,2001] Bespalova [2001] ha studiato cinque famiglie affette da sordità progressiva sulle frequenze gravi in Belgio, Olanda e Stati Uniti: nelle cinque famiglie la sordità era dovuta a una mutazione di WFS1. WFS1 è pertanto probabilmente un gene frequente in queste forme con curva audiometrica ascendente.
Non si conosce ancora la prevalenza del WFS1 nelle varie popolazioni e il suo possibile coinvolgimento in sordità dominanti con curve audiometriche non ascendenti. La diagnosi molecolare della WFS1 può essere proposta nelle famiglie con sordità autosomica dominante prevalente sulle frequenze gravi o a inizio su di esse.
Gene POU3F4
Il gene POU3F4 è stato il primo gene identificato responsabile di una ipoacusia non sindromica27. È un fattore di trascrizione, implicato nella morfogenesi. Esso è responsabile di una rara forma di ipoacusia, DFN3: la sordità mista legata all’X con geyser labirinto, per una rassegna, vedi [Cremers et al 2000] Il gap trasmissivo è importante, con Rinne di 50-60 dB, facendo sospettare un blocco ossiculare e portando a eseguire una timpanotomia esplorativa: ogni effrazione platinare provocherà un geyser massivo e porterà a una cofosi.
L’esame TC delle rocche petrose mostra in questa sindrome una dilatazione importante del condotto uditivo interno, una dilatazione cocleovestibolare e una scomparsa della segmentazione ossea cocleare.
Mutazioni del DNA Mitocondriale
Il DNA mitocondriale è una parte di DNA di piccole dimensioni situata in ogni mitocondri. Esso codifica per 13 acidi ribonucleici messaggeri (mRNA), due RNA ribosomiali (rRNA) e 22 RNA di trasporto ed è trasmesso, unicamente delle madri, a tutti i loro figli. Normalmente la maggior parte degli individui normali ha un solo tipo di DNA mitocondriale (omoplasmia), ma possono essere presenti diversi tipi di DNA mitocondriale per tessuto (eteroplasmia), il che spiega per esempio la variabilità dei fenotipi tra fratelli, che in teoria dovrebbero essere colpiti nella loro totalità.
Sono state descritte diverse mutazioni nelle sordità isolate: la prima descritta e la più frequente, la mutazione A1555G dell’RNA ribosomiale 12S, [80] quindi le mutazioni C1494T dell’RNA ribosomiale 12S e T7511C, T7510C dell’RNA di trasferimento della serina.
A1555G è particolarmente frequente in Spagna nelle famiglie affette da sordità con trasmissione compatibile con una sordità mitocondriale, fino al 25%. [81] La prevalenza è scarsa in altri paesi. [82] La sordità dovuta alla mutazione A1555G può essere di ogni grado e apparire a qualunque età, spontaneamente o dopo assunzione di aminoglicosidi. [83] In effetti il bersaglio degli aminoglicosidi è l’RNA ribosomiale batterico e la forma dell’RNA ribosomiale 12S con mutazione A 1555 G diviene più vicina agli rRNA batterici che legano gli aminoglicosidi con un’affinità anormalmente elevata. Questo spiega la comparsa di sordità per dosi normali di aminoglicosidi in questi pazienti. Anche l’altra mutazione dell’RNA ribosomiale 12SC1494T può indurre anche una suscettibilità agli aminoglicosidi. [84, 85]
IPOACUSIE SINDROMICHE PROGRESSIVE
La perdita progressiva dell’acuità uditiva è stata riscontrata in alcune sindromi caratterizzate da disturbi oculari, come la CHARGE, e la sindrome di Usher tipo III. Inoltre, la perdita dell’udito è progressiva nelle sindromi di Alstrom, Refsum e Norrie, nell’albinismo oculare, in alcuni tipi di atrofia otica, in alcune forme di cataratta. In quest’ultima condizione ereditaria, i risultati istopatologici hanno evidenziato una grave degenerazione cocleosacculare, Inoltre, nella sindrome di Stickler (o artro-oftalmopatia ereditaria), una perdita uditiva neurosensoriale e progressiva per le alte frequenze è riportata nelI’80% dei casi In genere si ha progressivo indebolimento dell’udito nella sindrome di Alport (nefrite e ipoacusia neurosensoriale), di Epstein (macrotrombocitopenia, nefrite e ipoacusia neurosensoriale), di Lemieux-Neemeh (nefrite, neuropatie motorie e sensitiva, ipoacusia progressiva) e di Muckle-WelIs (nefrite, orticaria, amiloidosi e ipoacusia neurosensoriale). Solitamente i pazienti presentano ipoacusia di grado severo ed il tasso di progressione non è correlato al grado di compromissione della funzionalità renale. La maggior parte delle ipoacusie neurosensoriali associate a disordini del sistema nervoso sono di fatto progressive. È così per le sindromi associate ad atassia (sindrome di Cockayne, di Klippel-Drante, di Hallgren, di Richards-Rundle), per quelle associate a disordini neuromuscolari (sindrome di Brown-Vialetto-Van Laere, la sindrome autosomica dominante di Charcot-Marie-Tooth, di Hagemoster, la sindrome di Pauli, ecc,) e per quelle associate a neuropatie sensoriali e del sistema nervoso autonomo (sindrome di Hicks, ecc), o sindromi mitocondriali come la sindrome di Kearns-Sayre (oftalmoplegia maggiore), MELAS (encefalomiopatia mitocondriale, acidosi lattica, episodi ictus-simile e ipoacusia neurosensoriale), o altre forme come la sindrome di Wells-Jankovic (paraplegia spastica, ipogonadismo e ipoacusia neurosensoriale), Poche anomalie dell’orecchio interno sono riportate da un punto di vista istopatologico’ atrofia del neuroepitelio, degenerazione delle cellule ciliate, grave degenerazione cocleosacculare, prominenza vascolare con emorragie e degenerazione quasi completa delle fibre nervose della lamina spirale, gliosi dei nuclei cocleari ventrali e degenerazione dei nervi cocleovestibolari
L’ipoacusia neurosensoriale a carico delle alte frequenze è un riscontro comune in età giovane-adulta nei disordini metabolici come l’alfa-D-mannosidosi, la malattia di Fabry, la malattia di Krabbe e l’adrenoleucodistrofia. Le mucopolisaccaridosi (MPS) sono un gruppo di disordini metabolici che coinvolgono il metabolismo degli eteroglicani, e ciascuna è caratterizzata dalla carenza di uno specifico enzima Le mucopolisaccaridosi sono prevalentemente malattie pediatriche La MPSII (malattia di Hunter), la forma lieve, è compatibile con la sopravvivenza fino all’età adulta, e un certo grado di perdita uditiva si sviluppa nella maggior parte delle persone interessate, il deficit è di solito misto e deriva da una combinazione di infezioni dell’orecchio medio e infiltrazione di glicosaminoglicani del ganglio otico La MPSIV (sindrome di Morquio) presenta ipoacusia neurosensoriale progressiva o mista che si sviluppa nella tarda infanzia Inoltre, nella sindrome DIDMQAD (diabete insipido, diabete mellito, atrofia otica, sordità neurosensoriale della sindrome di Wolfran), l’ipoacusia neurosensoriale coinvolge inizialmente le alte frequenze e si estende poi alle frequenze più basse, probabilmente correlata all’atrofia della stria vascolare
La maggior parte delle ipoacusie genetiche associate ad alterazioni dell’apparato tegumentario o a disturbi oro-dentali sono congenite e profonde, ma alcune si presentano con perdita progressiva dell’udito, come la sindrome di Waardenburg di tipo Il, la sindrome di Crandall, la sindrome otodentale, e, in alcuni casi, l’ittiosi La maggior parte degli audiolesi X-linked sono affetti da mutazioni nel gene PQU3F4 che causano o perdita progressiva dell’udito mista o severa ipoacusia neurosensoriale congenita.
Le sordità sindromiche rappresentano solo una piccola percentuale di sordità del bambino (10 – 15% circa) e una porzione poco conosciuta, probabilmente inferiore, di sordità dell’adulto. Sono state descritte diverse centinaia di sindromi con sordità (vedi Gorlin R.J.), e a tutt’oggi è stato identificato più di un centinaio di geni. È comunque importante conoscere e ricercare le principali sindromi, poiché la gestione e la valutazione eziologica saranno differenti da una sordità non sindromica. In ragione del grande numero di sindromi rare con sordità qualsiasi patologia malformativa nel bambino deve far effettuare un esame uditivo sistematico. Per le sordità sindromiche inoltre che comprendono una lesione malformativa craniofaciale, la sordità è molto spesso aggravata da un’otite cronica e s’impone un monitoraggio otologico regolare.
Abbiamo elencato nella Tabella 4 le sette sordità sindromiche che ci sembrano più importanti e che devono essere conosciute dai diversi specialisti che assumono in carico le sordità, a motivo della loro frequenza e/o della loro gravità potenziale. La Tabella 5 elenca in modo più completo le sindromi no eccezionali i cui geni sono stati identificati (per una rassegna esauriente dei geni clonati della sordità sindromica, vedi Marlin S.). Descriveremo qui le sette sindromi della Tabella 1.
1.10.1.1 Tre sindromi autosomiche recessive
Tre sindromi autosomiche recessive devono essere sistematicamente ricercate: le sindromi di Pendred e di Usher, entrambe frequenti, e la rara sindrome di Jerwell e Lange-Nielsen. Tutte queste sindromi hanno la particolarità di presentarsi a lungo come una sordità isolata, e solo una valutazione sistematica può permettere di individuarle precocemente.
Questa sindrome è stata descritta da oltre 100 anni. È riconosciuta dall’associazione di una sordità di origine cocleare, il più delle evolutiva, prelinguale o postlinguale precoce, con un disturbo del metabolismo dello iodio che si manifesta con un gozzo tiroideo. Essa si trasmette un modo autosomico recessivo. Il gene in causa, PDS (attualmente chiamato SLC26A4), è implicato sia nella sindrome di Pendred (Everett L.A.),che in una forma di sordità che rimane isolata: DFNB4(Li X.C., Usami S.). Il confine tra la sindrome di Pendred e la forma di sordità isolata DNFB4 è a volte difficile da definire poiché, all’interno di famiglie affette da Pendred, uno o più individui possono non sviluppare la lesione tiroidea(Van Hauwe P.). Noi descriveremo la forma DFNB4 nel paragrafo «Sordità non sindromiche».
La prevalenza della sindrome di Pendred è stimata a 7-10 cas/ 100.000 nascite. La sordità ha la particolarità, quando non è molto profonda fin dall’inizio, di evolvere per gradi di peggioramento improvviso, seguiti da un recupero in genere parziale. Queste fluttuazioni sono estremamente invalidanti e angosciose per il bambino.
L’età di comparsa del gozzo tiroideo è variabile, il più delle volte, nel corso del secondo decennio di vita (da 6 a 37 anni con età media 14,9 anni nella recente casistica di Blons sul territorio francese) e in questa stessa casistica era associato un ipotiroidismo nel 77% dei casi(Blons H.). La sindrome di Pendred si presenta quindi come una sordità isolata per molti anni.
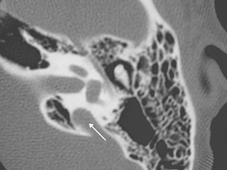
La TC delle rocche mette sempre in evidenza anomalie morfologiche dell’orecchio interno (dilatazione dell’acquedotto del vestibolo (Fig. 2 ), e a volte coclea incompleta e dilatata di tipo «Mondini») in modo quasi costante, ma queste anomalie possono essere monolaterali(Phelps P.D.).
|
|
|
Fig. 2 : Dilatazione dell’acquedotto del vestibolo (freccia) su una TC delle rocche in sezione assiale. |
Le anomalie di organificazione dello iodio possono essere messe in evidenza con la scintigrafia tiroidea con test al perclorato: l’incorporazione degli ioduri alla molecola di tireoglobulina avviene in modo anormale e la somministrazione di perclorato, anione inorganico, induce un rilascio di ioduri non organificati. La misura della quantità di iodio radioattivo prima e dopo somministrazione di perclorato mostra una diminuzione superiore al 10%. Questo test può permettere di individuare una sindrome di Pendred prima della comparsa di gozzo nei pazienti con malformazione dell’orecchio interno. Tuttavia questo tipo di test non è né sensibile (normale in certi soggetti in alcune forme familiari) né specifica (positivo in particolare nella tiroidite di Hashimoto e nei pazienti con mutazioni dei geni della tiroide perossidasi o della tiroglobulina). Ha inoltre l’inconveniente di sottoporre il paziente a irradiazione.
Il gene responsabile della sindrome di Pendred è stato localizzato nel 1997 nella regione 7q31( Coyle B., Sheffield V.C.), ed è stato clonato da Everett e coll. (Everett L.A.).Il gene PDS codifica per la pendrina formata da 780 aminoacidi. A tutt’oggi sono state identificate più di cinquanta mutazioni nel gene PDS. Queste mutazioni sono ripartite nei 21 esoni del gene. Quattro di esse sono particolarmente frequenti, presenti sul 74% dei cromosomi trasferiti (Blons H., Coyle B.,). Mutazioni della PD S sono evidenziate nella grande maggioranza dei pazienti che presentano una sindrome di Pendred in Francia ed è sempre presente una dilatazione dell’acquedotto del vestibolo nei soggetti in cui sono identificate le due mutazioni(Blons H.).
La sindrome di Usher associa alla sordità una retinite pigmentosa. Numerose mutazioni sono state identificate come responsabili di questa sindrome, che sulla base del grado di compromissione, dell’epoca di comparsa e dell’evoluzione delle patologie che la caratterizzano, è stata classificata in 3 diversi tipi diversi. I tre quarti sono Usher di tipo I . Nella sindrome di Usher tipo I sono stati identificati 7 sedi di linkage e 4 geni (MYO7A. USH1C. CDH23. PCDH15) a carico dei quali sono state osservate varie mutazioni, con sordità congenita profonda, areflessia vestibolare bilaterale responsabile di un ritardo della deambulazione (deambulazione dopo 18 mesi) e retinite che si sviluppa durante l’infanzia. I primi segni visivi sono dei disturbi della visione in penombra, spesso verso l’inizio del secondo decennio di vita, ma l’esame sistematico del fondo dell’occhio può consentire la diagnosi molto prima di questa età, fin dai 3-4 anni. L’esame più precoce è l’elettroretinogramma, patologico prima dei primi segni nel fondo dell’occhio. La sindrome di Usher di tipo I è un’indicazione all’impianto cocleare precoce per ottenere una comprensione del linguaggio senza lettura labiale in questi bambini che, in età adulta, avranno una rilevante compromissione visiva. Porre la diagnosi attraverso l’esame del fondo dell’occhio a 4 anni è quindi già una situazione tardiva. In linea di principio l’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi, e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale.
Condotta da tenere
L’esame oftalmologico esteso al fondo dell’occhio deve essere sistematico e ripetuto nel bambino e nell’adulto sordi e ogni sordità profonda congenita con ritardo della deambulazione senza eziologia evidente deve fare eseguire un elettroretinogramma, anche se il fondo dell’occhio è normale.
Nella sindrome di Usher tipo II sono state osservate varie mutazioni a carico dii gene (USH2A); tuttavia è verosimile che vi sia una eterogeneità genetica dal momento che sono state identificate 2 ulteriori sedi di linkage, la sordità è in media grave, non progressiva, predominante sulle frequenze acute, la retinite un poco più tardiva e i segni vestibolari assenti. Nella Usher tipo III è stato recentemente individuato 1 gene (USH3) a carico del quale sono state osservate varie mutazioni (Tab. I). la sordità è progressiva, i segni vestibolari e l’età di inizio della retinite sono variabili (per una rassegna, vedi [8]).
A tutt’oggi sono stati localizzati 11 geni e otto di questi geni sono stati identificati, cinque per la sindrome di Usher tipo I, due per il tipo II e uno per il tipo III (Tabella 4). MYO7A e CDH23 rappresentano rispettivamente il 30 e 29% dei casi di Usher tipo I e USH2A il 40% dei tipi II(Ouyang X.) La diagnosi molecolare non è eseguita di routine e la diagnosi è essenzialmente clinica.
Tab, II. Genetica della s, di Usher.
|
Locus |
Localizzazione |
Gene |
Marker dI screening |
Most lmportant Reference |
OMIM |
|
USH1A |
14q32 |
Unknown |
D14S250, D14S260, D14S292, D14S78 |
Kaplan et al., 1992 |
276900 |
|
USH1B |
11q135 |
MYO7A |
Di 1S906, Dl 1S911, D11S52, OMP-CA |
Weil et al,1995 |
276903 |
|
USH1C |
hp15.1 |
USH1C |
D11S902,D11S921, D11S899,D11S861 |
Smith et al,, 1992 |
276904 |
|
USH1D |
10q |
CDH23 |
D10S529, D10S202, D10S573 |
Wayne et al,, 1996 |
601067 |
|
USH1E |
21q |
Unknown |
D21S1884, |
Chaib et al, 1997 |
602097 |
|
USH1F |
10q2122 |
PCDH15 |
D10S199, D10S578, D10S596 |
Ahmed et aL, 2001 |
602083 |
|
USH1G |
17q24-25 |
Unknown |
|
Mustapha et al.., 2002 |
|
|
USH2A |
1q41 |
USH2A |
D1S229, D1S490, D1S237, D1S474 |
Kimberling et al, |
276901 |
|
USH2B |
3p2324..2, |
Unknown |
D3S1578, D3S3647, D3S3658 |
Hmani et al,, 1999 |
276905 |
|
USH2C |
5q14.3-q21.3 |
Unknown
|
D5S428, D5S421 |
Pieke-Dahl et al., 2000 |
605472 |
|
USH 3 |
3q21-q25 |
USH3 |
D3S1299, D3S1555, D3S1280, D3S1 279 |
Sankila et al, 1995 Joensuu et al., 2001 |
276902 |
Sindrome di Jerwell e Lange-Nielsen
È rara (1/100.000), ma di diagnosi facile tramite un elettrocardiogramma (ECG) sistematico in caso di sordità grave o profonda congenita(Splawski I.). L’ECG mostra un allungamento dello spazio QT che traduce un disturbo della conduzione cardiaca, fonte di malessere o di morte improvvisa che possono manifestarsi senza circostanze scatenanti o in seguito a uno stress. La prevenzione è possibile mediante un trattamento medico. Due geni sono stati identificati, KCNQ1 e KCNE1, (Splawski I., Neyroud N., Wang Z., Chen Q.) che codificano per alcuni canali del potassio localizzati nella stria vascolare (Tabella 4). I genitori portatori eterozigoti della mutazione possono presentare dei malesseri (donde l’importanza dell’anamnesi familiare) e un’ipoacusia.
1.10.1.1 Tre sindromi autosomiche dominanti
Devono essere conosciute dagli otorinolaringoiatri (ORL) poiché sono frequenti e probabilmente sottodiagnosticate: la sindrome di Waardernburg, la sindrome branchio-oto-renale (BOR) e la sindrome di Stickler.
la sindrome di Waardenburg è la causa più comune di perdita di udito sindromica autosomica dominante. Associa una sordità ad anomalie della pigmentazione dovute a un’assenza di melanociti in diversi organi. Ciò può riguardare i capelli (ciocche bianche) e le sopracciglia (Figura 3 ), gli occhi (occhi molto azzurri, depigmentati, depigmentazione sul fondo dell’occhio), la cute (macchie cutanee). In alcune forme (Waardernburg di tipo 1 e 3), è presente un’anomala distanza tra gli occhi, con i canti interni spostati verso l’esterno con riduzione di lunghezza della fessura palpebrale (distopia cantale) (Figura 3). La sordità è molto variabile, mono- o bilaterale, da leggera a profonda (profonda nel 35% dei casi), (Hildesheimer M.), con o senza malformazione dell’orecchio interno.

Figura 3 : Sindrome di Waardernburg di tipo I con distopia cantale (doppia freccia), occhi molto azzurri, sopracciglia parzialmente bianche.
La trappola per la diagnosi è che le particolarità fisiche che possono essere presenti nella famiglia dei sordi ed essere molto suggestive non sono spontaneamente descritte all’anamnesi, non essendo considerate patologie. Le ciocche bianche sono inoltre il più delle volte tinte. La domanda circa la presenza di occhi depigmentati o di ciocche bianche nella famiglia deve quindi essere posta sistematicamente. Sono stati descritti quattro tipi clinici di sindrome di Waardernburg in funzione dei segni associati:
• il tipo I è associato a una distopia cantale ed è causata da mutazioni in PAX3.;
• il tipo II, il più frequente, è privo di distopia cantale canti ed è causata da mutazioni in MITF.;
• il tipo III (o Klein-Waardenburg) è un tipo I associato a malformazioni degli arti superiori ed è causata da mutazioni in PAX3.;
• il tipo IV è un tipo II con malattia di Hirschsprung. Attualmente, sono stati identificati sei geni, onnicomprensivi nella Tabella 4. Essi non rendono conto di tutte le sindromi di Waardernburg (per esempio, PAX3 è chiamato in causa in tre quarti dei tipi I e MITF nel 15% dei tipi II soltanto) (Tassabehji M.).
La sindrome branchio-oto-renale (BOR) è la seconda causa più comune di perdita di udito sindromica autosomica dominante ,.associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000(Fraser F.C.). Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un’interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un’ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell’orecchio esterno (malformazioni del padiglione, aplasia dell’orecchio, encondromi, stenosi dei condotti uditivi) (Figura 4 ), dell’orecchio medio (esiste una componente di trasmissione all’audiogramma) e dell’orecchio interno (varie malformazioni cocleovestibolari) (Figura 5 ). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi (Figura 6 ). In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell’orecchio esterno, è consigliabile eseguire un’ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1(Abdelhak S., Ruf R.G.). Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale(Vincent C.).
|
|
|
Fig. 4 : Orecchie a trombetta ed encondromi. |
|
|
|
Fig. 5 : Micrococlea in una sindrome branchio-oto-renale (BOR). |
|
|
|
Fig. 6 : Residuo branchiale cervicale. |



Questa sindrome è dovuta a una alterazione delle catene ![]() di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
La sordità di percezione, trasmissione o mista è incostante, e spesso mascherata o aggravata dai problemi di otite cronica che sono associati all’incompetenza faringea. Essa è spesso evolutiva. Sono descritti tre tipi di sindrome di Stickler: nei tipi 1 e 3, agli altri elementi della sindrome è associata una miopia molto forte con rischio di degenerazione vitro-retinica. L’esame oftalmologico di ogni bambino sordo e di ogni lattante con incompetenza faringolaringea deve permettere d’individuare questa sindrome e correggere precocemente la miopia.
L’otosclerosi è una malattia genetica generalmente associata ad insorgenza nell’età adulta ipoacusia trasmissiva. Tuttavia, otosclerosi avanzata può provocare SNHL. I geni responsabili otosclerosis non sono state trovate, ma foci sui cromosomi 6, 7, e 15 sono stati implicati.
l’acondroplasia può essere associato con la perdita uditiva mista.
La Malattia di Paget può causare progressive, ad insorgenza nell’età adulta ipoacusia trasmissiva, sordità neurosensoriale genetica (SNHL), o entrambi. Altri reperti comuni di questo disturbo osseo sono l’allargamento del cranio, cifosi, e un accorciamento della statura. La perdita dell’udito è probabilmente dovuto ad un processo cocleare. Fattori genetici e ambientali sono suscettibili di essere fattori che contribuiscono.
1.10.1.2 Sindrome legata al cromosoma X
Una sindrome legata al cromosoma X, b)la sindrome di Alport, deve essere sistematicamente ricercata in ogni sordità postlinguale e/o evolutiva. La sordità è progressiva (1o decennio) associata a episodi di ematuria (l’ematuria inizialmente microscopica per diversi anni) con un interessamento renale che evolve verso l’insufficienza renale. Il controllo oftalmologico riscontra un lenticono anteriore e una cataratta polare anteriore, suggestivi di questa sindrome.
Per le sindromi di Alport dominanti legate al cromosoma X, dovute a mutazioni del gene COL4A5, sono affetti uomini e donne, ma la lesione è più moderata nelle donne. La sordità può precedere la lesione renale e interessa a 40 anni 90% degli uomini e 10% delle donne. A 40 anni, 90% degli uomini e 12% delle donne è colpito da insufficienza renale(Jais J.P.). Lo stick urinario sistematico nel bambino sordo permette una diagnosi e una gestione precoci della sindrome di Alport.
Diversi geni sono identificati per questa sindrome (Tabella 4). Essi codificano collageni di tipo IV che entrano nella composizione delle membrane basali renali, cocleari e della capsula anteriore del cristallino.
Esistono anche delle sindromi di Alport a trasmissione autosomica recessiva, nelle quali le femmine presentano la stessa lesione dei maschi. Le trasmissioni autosomiche dominanti sono infine possibili ma molto rare.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||
|
|
Didascalia : |
SORDITÀ NON SINDROMICHE approfondimento
Le forme autosomiche recessive sono le più frequenti e la sordità è in generale congenita. Nelle forme dominanti, la sordità è il più delle volte progressiva o a comparsa ritardata, nel corso dell’infanzia o in età adulta. Noi affronteremo qui solo le forme più frequenti. [Anche se in letteratura sono riportati molti casi di famiglie con mutazione autosomica dominante (AD) che hanno manifestato forme d’ipoacusia progressiva non esistono dati epidemiologici sulla prevalenza delle ipoacusie genetiche nella popolazione adulta. La DFNA1 è la tipica forma (ipoacusia di ‘Monge’) in cui sono coinvolte le basse frequenze durante i primi anni, si ha la progressione della compromissione dell’udito che comporta il coinvolgimento delle alte frequenze negli anni successivi. Individui più anziani con questo genotipo mostrano una perdita uditiva che coinvolge tutte le frequenze. Questo vale anche per alcuni altri disordini non sindromici dell’udito che all’inizio possono avere differenti profili audiometrici (ad esempio, che coinvolgono medie o alte frequenze) che diventano sovrapponibili in età adulta, ad esempio grave ipoacusia pantonale con possibile caduta sulle alte frequenze Si definisce “ipoacusia progressiva” una perdita di 15 dBHL nel PTA in un periodo di 10 anni (European Work Group on Genetic Hearing lmpairment, 2000).
Studi recenti hanno identificato tre possibili geni responsabili di ipoacusia neurosensoriale progressiva a trasmissione autosomico-recessiva LOXHD1 (lipoxygenase homology domains 1), PJVK (pejvakin) e MYO3A (miosina 3A), espressi nelle cellule ciliate dell’orecchio interno ed in particolare PJVK (che codifica per una proteina citoplasmatica) è espresso a livello del ganglio spirale Questi tre geni sono indispensabili per la normale fisiologia delle cellule ciliate dell’orecchio interno Nei modelli animali, mutazioni a carico del gene LOXHD1 alterano la normale funzionalità delle cellule ciliate; studi condotti sul DNA di famiglie affette da ipoacusia progressiva hanno permesso di individuare una mutazione responsabile di DFNB77, a trasmissione autosomica recessiva, e il gene umano corrispondente è stato localizzato sul braccio lungo del cromosoma 18 (Grillet et al .., 2009). Altre forme progressive autosomico-recessive sono attribuite a mutazioni dei geni PVJK e MYO3A: rispettivamente DFNB59, caratterizzata da neuropatia (Delmaghani et al.., 2006) e DN FB3O..
Altri geni le cui mutazioni possono determinare ipoacusia neurosensoriale progressiva sono MYH9 (Myosin, heavy chain 9) e WFS1 (sindrome di Wolfram o DIDMOAD) In particolare mutazioni a carico del gene MYH9, che codifica per la catena pesante della miosina non-muscolare lIA, sono responsabili di fenotipi riconducibili alla sindrome di Epstein, di Fechtner, di Sebastian e all’anomalia di May-Hegglin. Tale classificazione è da ritenersi tuttavia superata perchè non in grado di descrivere la complessità dei quadri fenotipici conseguenti alle mutazioni del gene A quadri cImici così ampi e variabili è stato dato il nome di disordini MYH9-correlati (o malattia MYH-9 correlata), caratterizzati da grandi piastrine e in bassa concentrazione, glomerulonefrite e ipoacusia neurosensoriale progressiva per le alte frequenze che insorge in età giovane adulta, Tutti i pazienti presentano alla nascita piastrinopenia, ma alcuni di loro sono destinati a sviluppare negli anni successivi glomerulonefrite cronica e/o deficit uditivo neurosensoriale e/o cataratta presenile Il pattern clinico è ereditato in maniera autosomica dominante, A trasmissione autosomica recessiva è invece la sindrome di Wolfram (o DIDMOAD, nota anche come DFNA 6/14/38), dovuta a mutazioni a carico del gene WFS1, caratterizzata da una perdita progressiva sulle basse frequenze, dopo un iniziale decadimento sui toni acuti, disordini neurodegenerativi progressivi, diabete mellito e atrofia ottica prima dei 15 anni, atassia, neuropatia periferica e altre alterazioni neurologiche] DA MARTINI A. e PROSSER S.”AUDIOLOGIA E FONIATRIA” 2011 ed omega . Le Tabella 1, Tabella 2, Tabella 3 analizzano i loci e i geni identificati al 1o ottobre 2004 (per l’aggiornamento dei dati e un elenco accurato dei diversi loci e geni noti, verificare il sito Hereditary Hearing Loss Homepage(Van Camp G.).
Le connexine sono piccole proteine transmembranarie, coinvolte nella formazione di giunzioni comunicanti tra cellule adiacenti, per la maggior parte delle cellule dell’organismo.
Attualmente nei mammiferi sono note cinque connexine diverse. Quattro geni di connexine, espressi nell’orecchio interno, possono essere implicati in sordità isolate o sindromiche: GJB2 (connexina 26, in 13q11.q12), GJB6 (connexina 30, in13q11.q12), GJB3 (connexina 32, in Xq13.1) e GJB1 (connexina 31, in 1p34) (Tabella 1, Tabella 2) (per un rassegna vedere(Denoyelle F.). GJB2 è conosciuto fin dal 1997 come responsabile della forma principale di sordità del bambino, DFNB1,( Zelante L.) e, più recentemente, tre equipe hanno dimostrato che, in modo abbastanza frequente, una sordità DFNB1 poteva essere dovuta all’associazione di una delezione di GJB6, situato nello stesso locus del GJB2, con una mutazione di GJB2 (vedi i capitoli seguenti).
Si riteneva inizialmente che le connexine 26 e 30 fossero coinvolte nel ricircolo del potassio nell’endolinfa e che la sordità potesse essere dovuta a un’alterazione della concentrazione di K+ endolinfatico. La patogenesi della sordità è meglio conosciuta da quando sono disponibili modelli animali di delezione parziale o totale dei geni GJB2 (Cx26) et GJB6 (Cx30): l’orecchio interno si sviluppa normalmente, e una morte cellulare, inizialmente a livello delle cellule di sostegno delle cellule ciliate interne, sopraggiunge verso il 14o giorno postnatale nel topo. La concentrazione di potassio nell’endolinfa rimane normale fino a questa data. La morte cellulare si estende poi alle altre cellule di sostegno e alle cellule ciliate, facendo avanzare l’ipotesi di una tossicità scatenata dalla risposta delle cellule ciliate interne alla stimolazione sonora, per esempio un mancato ricircolo del glutammato(Cohen-Salmon M.).
Gene della connexina 26 (CX26 o GJB2)
GJB2 è chiamato in causa da un lato nella forma più comune di sordità del bambino, la sordità autosomica recessiva DFNB1 e, dall’altro, in una forma rara di sordità autosomica dominante, DFNA3(Kelsell D.P., Denoyelle F.). Mutazioni particolari di questo gene rendono conto anche di tre sindromi rare che associano sordità e interessamento cutaneo, la sindrome di Vohwinkel, la sordità con cheratosi palmoplantare e la sindrome Keratosis Ichthyosis Deafness (KID)( Maestrini E., Kelsell D.P., Richard G.) . Dal 1997 si sa che la forma DFNB1 è in causa nella metà delle sordità recessive congenite e 30 – 40% dei casi sporadici congeniti in Francia(Zelante L.). A causa del baso numero di figli per famiglia in Francia, molte delle sordità dovute a mutazioni del GJB2 si presentano in effetti come casi isolati. In questa forma di sordità DFNB1, i soggetti sordi portano delle mutazioni sui due alleli del gene, mentre i soggetti eterozigoti sono normoudenti. La deficienza uditiva è congenita e stabile nella maggioranza dei casi (un’evolutività è osservata in meno del 20% dei casi, il più delle volte lieve). Non è stato descritto alcun episodio di aggravamento improvviso della sordità in questa forma. La sordità può presentarsi in tutte le gradazioni, ma è più spesso profonda (vedi più avanti la gravità in funzione del tipo di mutazioni). Le curve audiometriche sono, nella maggior parte dei casi, piatte (lesione simile di tutte le frequenze) o discendenti (interessamento preferenziale delle frequenze acute). La TC delle rocche e le prove vestibolari caloriche sono normali(Denoyelle F.).
In Francia, come nei paesi occidentali e mediterranei, predomina ampiamente una mutazione: 35delG. La mutazione 35delG è dovuta alla delezione di una base di DNA, una guanina, in posizione 35 nella parte codificante del gene. Questa mutazione determina un ritardo del quadro di lettura e porta alla formazione di una proteina tronca.
35delG rappresenta il 58 – 67% degli alleli mutati in Francia(Roux A.F., Marlin S.), 55 – 66% in Spagna e Italia, 46% negli Stati Uniti(Cryns K.). I portatori eterozigoti (che portano la mutazione su un singolo allele del gene e sono normoudenti) di questa mutazione sono molto frequenti nella popolazione generale, tra il 2,5 e il 4% della popolazione in Francia, Spagna e Italia, 2 % negli Stati Uniti(Roux A.F., Gasparini P.). La prevalenza delle mutazioni di GJB2 è simile in molti paesi a quella del gene CFTR della fibrosi cistica. Una diagnosi molecolare di routine è disponibile in molti laboratori in Francia.
Altre mutazioni maggioritarie sono riscontrate in alcune popolazioni: 167delT nella popolazione ebrea ashkenazita(Morell R.J.), 235 delC in Giappone, in Cina e in Corea(Fuse Y., Kudo T.) , R143W nell’Africa nera (mutazione la cui patogenicità è attualmente rimessa in causa)( Brobby G.W.). In Francia sono state descritte una trentina di mutazioni (60 nel mondo) e alcune sono ricorrenti: 310 del14, E47X, L90P, Q57X, rappresentano tra il 2 e il 3% degli alleli mutati. Si noti la presenza di una delezione frequente del gene della connexina 30, situato nell’intervallo del locus DFNB1, e che rappresenta la mutazione del secondo allele in associazione con una mutazione del GJB2 nel 5% dei casi (vedi oltre nel capitolo «Altri geni di connexina»). Le mutazioni “inattivanti” che portano a una proteina tronca sono associate a sordità più gravi delle mutazioni “non inattivanti”, del tipo falso senso(Marlin S., Cryns K.). In particolare la mutazione frequente L90P associata a 35delG sull’altro allele, (genotipo L90P/35delG) è più comunemente associata a una sordità leggera media rispetto al genotipo 35delG/35delG(Marlin S.).
Dopo molteplici controversie la mutazione molto frequente M34T, considerata inizialmente come una mutazione dominante quindi recessiva, è forse non patogena nella maggior parte dei casi: sono stati segnalati molti soggetti 35delG/M34T normoudenti(Gasparini P.) e questa variante è altrettanto frequente nella popolazione sorda che nella popolazione generale (frequenza allelica di 1,6% contro 1,15%( Roux A.F.). Molti altri polimorfismi del gene della connexina 26 sono stati identificati a tutt’oggi.
La messa in evidenza di mutazioni patogene sui due alleli del GJB2 in presenza di un caso sporadico di sordità congenita permette di affermare il carattere genetico della sordità e segnalare alle famiglie il rischio di nuovi casi. In alcune forme familiari la diagnosi molecolare di mutazioni del GJB2 può permettere di precisare una modalità di trasmissione poco chiara.
Connexina 26 e sordità autosomica dominante
Il coinvolgimento del gene della connexina 26 nella forma di sordità DFNA3 è stata a lungo oggetto di controversie, in quanto la mutazione inizialmente descritta, M34T(Kelsell D.P.) era considerata di dubbia patogenicità. In seguito sono state segnalate due mutazioni chiaramente patogene del gene della connexina 26 (W44C, C 202 F) in tre grandi famiglie lionesi(Denoyelle F., Morell R.J.), e sono poi state segnalate altre mutazioni. Le caratteristiche della sordità, trasmessa in tutti i casi in modalità autosomica dominante, sono molto diverse a seconda delle mutazioni: per esempio, per W44C la sordità è prelinguale, da media a profonda e colpisce tutte le frequenze, mentre per C 202 F la sordità compare tra 10 e 20 anni, colpisce inizialmente le alte frequenze ed evolve lentamente per divenire leggera-media a 50 anni(Feldmann D., Bozon M.).
Altri geni delle connexine possono essere responsabili di una sordità isolata, autosomica recessiva e/o dominante (Tabella 1, Tabella 2): GJB6 e GJB3 che codificano rispettivamente per le connexine 30 e 31. Sono implicati in sordità sindromiche ma non in sordità isolate: GJB1, il gene della connexina 32, responsabile di una forma della sindrome di Charcot-Marie-Tooth nella quale può essere presente una sordità(); [Stojkovic T.] GJA1 (connexina 43 ), (descritto come responsabile di sordità isolata, fatto in seguito smentito), coinvolto in una sordità sindromica rara con sordità di trasmissione, la displasia oculo-dento-digitale(). [Paznekas W.A.]
Il gene GJB6 (connexina 30) è particolarmente importante: molto simile al gene della connexina 26, una delezione di un singolo allele di questo gene (GJB6 -D13S1830del), è frequentemente riscontrata in associazione con una mutazione eterozigote del gene della connexina 26 in soggetti sordi congeniti() [del Castillo I.; Lerer I.;Pallares-Ruiz N.] (i soggetti sordi sono quindi portatori di una mutazione patogena di un allele del gene della connexina 26 e la delezione su un allele del gene della connexina 30). La conoscenza di questa delezione ha permesso di provare il carattere genetico della sordità in alcuni soggetti che presentano una sola mutazione del gene della connexina 26 (6% dei casi in una coorte francese di 255 bambini sordi con sordità congenita di ogni grado senza malformazioni dell’orecchio interno)(). [48]
Punto essenziale
GJB2, gene della connexina 26, rende conto di più di un terzo delle sordità congenite in Francia
La sordità dovuta ai geni delle connexine 26 e 30 (geni GJB2, GJB6 ) è congenita bilaterale isolata (diagnostica per immagini normale, assenza di disturbi vestibolari), il più delle volte profonda. La diagnosi molecolare è disponibile per questi due geni: prima diagnosi molecolare richiesta dal genetista nelle sordità prelinguali, in assenza di malformazioni dell’orecchio interno o se la diagnostica per immagini cerebrale non è eseguita a causa della giovane età.
Indicazioni della diagnosi molecolare dei geni delle connexine 26 e 30 (GJB2, GJB6)
In presenza di una sordità non sindromica prelinguale del bambino, la diagnosi molecolare di mutazioni del GJB2/GJB6 è la prima a essere proposta al termine della valutazione eziologica se la diagnostica per immagini della rocca petrosa è normale o se non ha potuto essere realizzata a causa della giovane età del paziente.
Nell’adulto bisogna pensare a chiedere queste diagnosi molecolari quando la sordità risale all’infanzia e diffidare di altre cause sovrapposte che hanno potuto aggravare la sordità (esposizione al rumore, otosclerosi, otiti croniche ecc.).
Questi due geni delle connexine sono di piccole dimensioni, composti da un esone codificante e un esone non codificante. Per il gene GJB2 l’analisi avviene abitualmente con sequenziamento diretto o con cromatografia ad alta prestazione (DHPLC). La ricerca della delezione (GJB6-D13S1830) è basata sul fatto che questa delezione comporta un sito di restrizione.
Nella patologia recessiva ricercata DFNB1 devono essere mutati i due alleli del gene GJB2 (o un allele del GJB2 e uno del GJB6) per affermare che si tratti effettivamente di questa forma di sordità. La strategia abituale è ricercare fin dall’inizio delle mutazioni nell’esone codificante del GJB2. In assenza di mutazioni scoperte o se è rilevata solo una mutazione la ricerca di mutazioni è condotta nell’esone non codificante del GJB2 ed è ricercata la delezione di GJB6.
La messa in evidenza di mutazioni patogene sui due alleli del GJB2 e/o GJB6 in presenza di un caso sporadico di sordità congenita permette di affermare il carattere genetico della sordità e segnalare alle famiglie il rischio di nuovi casi, 25%, nei fratelli. In alcune forme familiari la diagnosi molecolare può permettere di precisare una modalità di trasmissione poco chiara.
1.10.2.2 Sordità isolata dovuta al gene PDS, DFNB4
Il gene della sindrome di Pendred, PDS, può essere responsabile della forma di sordità DFNB4, che ricorda in ogni punto quella della sindrome (prelinguale, progressiva o fluttuante, malformazione dell’orecchio interno), ma che resta isolata, cioè senza interessamento tiroideo. Nel 1998 e 1999 due studi hanno dimostrato il coinvolgimento di mutazioni del PDS nella forma di sordità DFNB4, in soggetti che presentano una sordità congenita profonda neurosensoriale senza gozzo né ipotiroidismo, ma con una malformazione dell’orecchio interno alla TC delle rocche (dilatazione bilaterale dell’acquedotto del vestibolo [DAV], che contiene il sacco endolinfatico, in cui il criterio è rappresentato da un acquedotto più largo di 2 mm nella parte mediana)( Li X.C., Usami S.). Il test al perclorato era normale nei soggetti testati.
Campbell et al.( 2001) hanno mostrato che PDS era molto frequentemente coinvolto in alcune forme familiari di sordità con malformazioni dell’orecchio interno: mutazioni di questo gene sono messe in evidenza in quattro famiglie su cinque con malformazione di Mondini e cinque famiglie su sei con DAV. In una casistica francese molto recente le mutazioni del PDS sono riscontrate nel 42% dei pazienti con sordità e malformazione dell’orecchio interno (in quasi la metà dei casi, si è potuta identificare solo una mutazione), ed era sempre presente una DAV nei pazienti con mutazione doppia(Albert S.). Il riconoscimento di sordità dovute alla PDS è importante per la consulenza genetica ma anche prognostica, poiché prevedere le fluttuazioni e l’evolutività può aiutare l’orientamento educativo e far prendere delle precauzioni nei confronti dei microtraumi (alcuni sport violenti, barotraumi) in questi bambini. Il trattamento degli episodi di aggravamento della sordità non è ben codificato e si basa spesso su un trattamento con vasodilatatori/corticosteroidi come si usa nella sordità improvvisa, benché questo non sia validato, ma bisogna sicuramente evitare i ricoveri iterativi con le loro ripercussioni psicologiche e scolastiche per questi bambini il cui stato può fluttuare in tempi molto ravvicinati.
1.10.2.3 Gene dell’otoferlina, OTOF
Le mutazioni del gene dell’otoferlina, OTOF, sono responsabili della forma di sordità recessiva DFNB9(Yasunaga S.). L’otoferlina sarebbe implicata nel traffico vescicolare presinaptico delle cellule ciliate interne(Petit C.). La sordità è grave o profonda, prelinguale, e una mutazione di questo gene, Q829X, sembra particolarmente frequente in Spagna(Migliosi V.). La particolarità di questa forma di sordità è che essa può presentarsi come una neuropatia auditiva, con otoemissioni acustiche inizialmente conservate. Starr(1996) ha per primo utilizzato il termine di neuropatia uditiva per designare nell’adulto delle sordità, spesso bilaterali, associate a un’alterazione dei potenziali evocati uditivi (PEU) contrastante con la presenza di otoemissioni acustiche (OEA) normali. Queste forme sono oggi meglio conosciute nell’adulto e nel bambino e giustificano la valutazione delle otoemissioni per individualizzarle nel corso della gestione di una sordità di percezione.
La frequenza di queste sordità è molto elevata (1%) nei neonati ricoverati in terapia intensiva, il che giustifica uno screening delle sordità con i potenziali evocati piuttosto che con OEA in questa popolazione. Le mutazioni dell’otoferlina sono probabilmente rare tra i bambini con una diagnosi di «neuropatia auditiva»: Madden, (2002) tra 22 casi pediatrici, descriveva 15 casi (68%) con patologie neonatali intricate (iperbilirubinemia: 11/15, prematurità: 10/15, farmaci ototossici: 9/15, ventilazione assistita: 8/15) e e sei casi erano rappresentati da coppie di due fratelli sordi che evocavano una causa genetica autosomica recessiva, non essendo stato ricercato il gene della DFNB9, OTOF .
Il termine neuropatia uditiva sembra assolutamente inappropriato per la sordità dovuta al gene OTOF, sordità di origine cocleare per la quale i risultati di impianto cocleare sembrano del tutto paragonabili ai risultati abituali dell’impianto(Loundon N.).
Questo gene è espresso soprattutto nelle cellule ciliate esterne e codifica per un canale del potassio voltaggio-dipendente. Esso è coinvolto in una forma di sordità progressiva, DFNA2, che interessa inizialmente le frequenze acute. L’età di esordio è variabile, tra 1 e 30 anni, la sordità progredisce di circa 1 dB per anno, e gli acufeni sono frequenti(Coucke P.J., Kubisch C.).
La sordità dovuta alla lesione del COCH, DFNA9, ha inizio sulle frequenze acute nell’adolescenza o nell’adulto e progredisce rapidamente per interessare tutte le frequenze. L’evoluzione è spesso asimmetrica, con perdita media tra 3 e 4 dB per anno(Verhagen W.I.). La sordità può essere grave a profonda a partire da 60 anni.
Alcuni pazienti hanno episodi di vertigini, sensazione di «pienezza auricolare» e acufeni che possono evocare una malattia di Ménière, anche se la forma della curva è diversa(Robertson N.G.,). Diversi studi tra pazienti affetti della malattia di Ménière non hanno però riscontrato mutazioni del COCH.
Questo gene è espresso in grande quantità nella coclea. Esso codifica per una proteina della matrice extracellulare cocleare, la coclina. Dal punto di vista istologico si ritrovano, in questa forma di sordità, dei depositi acidofili cocleari molto caratteristici.
1.10.2.6 Gene della wolframina, WFS1
Questo gene è stato identificato nel 2001 come responsabile di una sordità sindromica, la sindrome di Wolfram, nella quale la sordità è associata a diabete mellito e insipido e a sintomi neurologici e oculari(Khanim F). Due equipe hanno dimostrato che WFS1 era anche in causa in una forma non sindromica autosomica dominante, DFNA38, caratterizzata da una sordità progressiva, che inizia tra 5 e 15 anni e colpisce di preferenza le frequenze gravi(Bespalova I.N., Young T.L.). La sordità colpisce in seguito tutte le frequenze e diventa media/grave verso i 40 anni. I loci DFNA6, DFNA14 e DFNA38 si sovrappongono e corrispondono tutti al gene WFS1. Young et al. hanno messo in evidenza mutazioni in due famiglie statunitensi e due famiglie olandesi in cui la sordità era legata al DFNA38, ma anche in due famiglie testate senza precedente localizzazione, con sordità dominante che interessava le basse frequenze(Young T.L.). Bespalova (2001) ha studiato cinque famiglie affette da sordità progressiva sulle frequenze gravi in Belgio, Olanda e Stati Uniti: nelle cinque famiglie la sordità era dovuta a una mutazione di WFS1. WFS1 è pertanto probabilmente un gene frequente in queste forme con curva audiometrica ascendente.
Non si conosce ancora la prevalenza del WFS1 nelle varie popolazioni e il suo possibile coinvolgimento in sordità dominanti con curve audiometriche non ascendenti. La diagnosi molecolare della WFS1 può essere proposta nelle famiglie con sordità autosomica dominante prevalente sulle frequenze gravi o a inizio su di esse.
Il gene POU3F4 è stato il primo gene di sordità non sindromica identificato(de Kok Y.J.). È un fattore di trascrizione, implicato nella morfogenesi. Esso è responsabile di una rara forma di sordità, DFN3, particolarmente importante da conoscere, in particolare per gli otorinolaringoiatri: la sordità mista legata all’X con geyser-labirinto. La sordità di percezione si accompagna a un aspetto trasmissivo importante, con Rinne di 50-60 dB (Figura 9), che ha spesso, prima che questa sindrome sia nota, fatto sospettare un blocco ossiculare portando a eseguire un esame dell’orecchio: ogni effrazione platinare provoca un geyser massivo e porta a una cofosi. Si sa attualmente che ogni intervento chirurgico per sordità di trasmissione a timpano normale nel bambino deve essere preceduto da una TC delle rocche, che mostra in questa sindrome una dilatazione importante del condotto uditivo interno, una dilatazione cocleovestibolare e una scomparsa della segmentazione ossea cocleare, con il modiolo che non è più evidente ala TC (Figura 10) (per una rassegna, vedi(Cremers F.P.). Le mutazioni del POU3F4 non sono rilevate in tutti i pazienti con questo fenotipo ed è possibile che siano implicati uno o più altri geni(Friedman R.A.). La vaccinazione antipneumococcica e, nel bambino piccolo, anti- Haemophilus influenzae deve essere consigliata nei pazienti con fenotipo clinico DFN3.
|
|
|
Fig. 10 :Sordità mista legata al cromosoma X DFN3: TC delle rocche in sezione assiale (A) e coronale (B) con dilatazione del condotto uditivo interno (CUI) e della coclea e scomparsa del modiolo osseo. |

1.10.2.8 Mutazioni del DNA mitocondriale implicate nei casi di sordità non sindromiche
Il DNA mitocondriale è una parte di DNA di piccole dimensioni situata in ogni mitocondri. Esso codifica per 13 acidi ribonucleici messaggeri (mRNA), due RNA ribosomiali (rRNA) e 22 RNA di trasporto ed è trasmesso, unicamente delle madri, a tutti i loro figli. Normalmente la maggior parte degli individui normali ha un solo tipo di DNA mitocondriale (omoplasmia), ma possono essere presenti diversi tipi di DNA mitocondriale per tessuto (eteroplasmia), il che spiega per esempio la variabilità dei fenotipi tra fratelli, che in teoria dovrebbero essere colpiti nella loro totalità.
Sono state descritte diverse mutazioni nelle sordità isolate: la prima descritta e la più frequente, la mutazione A1555G dell’RNA ribosomiale 12S(Prezant T.R.), quindi le mutazioni C1494T dell’RNA ribosomiale 12S e T7511C, T7510C dell’RNA di trasferimento della serina.
A1555G è particolarmente frequente in Spagna nelle famiglie affette da sordità con trasmissione compatibile con una sordità mitocondriale, fino al 25%(Estivill X.). La prevalenza è scarsa in altri paesi(Fichel-Ghodsian N.)). La sordità dovuta alla mutazione A1555G può essere di ogni grado e apparire a qualunque età, spontaneamente o dopo assunzione di aminoglicosidi(El-Schahawi M.). In effetti il bersaglio degli aminoglicosidi è l’RNA ribosomiale batterico e la forma dell’RNA ribosomiale 12S con mutazione A 1555 G diviene più vicina agli rRNA batterici che legano gli aminoglicosidi con un’affinità anormalmente elevata. Questo spiega la comparsa di sordità per dosi normali di aminoglicosidi in questi pazienti. Anche l’altra mutazione dell’RNA ribosomiale 12SC1494T può indurre anche una suscettibilità agli aminoglicosidi(Zhao H., Yoshida M.,).
1.10.4.2 ASindrome di Usher
È una patologia trasmessa con modalità autosomica recessiva: i portatori sono stimati essere tra 1/75 e 1/150 nella popolazione.
La sindrome di Usher è stata descritta nel 1858 da Von Grafe. il quale fu il primo ad associare r• •te pigmentosa e sordità neurosensoriale in un unica patologia: prese successivamente. Nel 1959. lo studioso Hallgren scoprì l’ereditarietà di questa sindrome.
E’ un chiaro esempio di affezione ereditaria di natura degenerativa. espressione di un alterazione nel metabolismo intracellulare già presente allo stadio embrionale, Caratterizzata clinicamente da ipoacusia neurosensoriale e rete pigmentosa. viene classificata in 3
diversi tipi sulla base delle caratteristiche cliniche (Tab. II)
I soggetti affetti presentano deficit uditivi e visivi, che possono comparire in epoche diverse nella vita e avere grado ed evoluzioni differenti secondo il tipo.
La rinite pigmentosa è una condizione in cui a livello di entrambi gli occhi si manifesta una degenerazione dei fotorecettori (bastoncelli) che hanno la funzione di reagire prevalentemente al contrasto tra chiaro e scuro e al movimento di oggetti. Questa degenerazione delle cellule fotorecettrici che progressivamente lasciano il posto ad accumuli di pigmento provoca dapprima una difficoltà a vedere in ambienti scarsamente illuminati, successivamente quali- do la degenerazione interessa oltre la parte più periferica anche la parte centrale della retina. si avrà un restringimento progressivo del campo visivo che potrà portare nel tempo a cecità totale. In particolare. nei soggetti affetti da sindrome di Usher Tipo I la retinite pigmentosa ha esordio molto precoce e spesso porta alla cecità: in quelli con sindrome di Usher Tipo II : l’ esordio della retinite pigmentosa avviene fra 10 e 20 anni e solitamente il quadro è meno grave rispetto al tipo I infine nella sindrome di Usher Tipo III i pazienti presentano un quadro variabile caratterizzato da progressivo peggioramento (Fishman. 1979: Fishman et al.. 1983)
L’ipoacusia è congenita. di tipo neurosensoriale. ha carattere prettamente cocleare ed è causata da malformazioni a carico dell’orecchio interno (01). Nella maggior parte dei pazienti con sindrome di Usher Tipo I l’ipoacusia è profonda. nella sindrome di Usher Tipo II solitamente l’ipoacusia è moderata con audiogramma in discesa sulle frequenze acute infine nella sindrome di Usher Tipo III l’ipoacusia è simile a quella descritta per il tipo II ,ma compare attorno ai 3-5 anni e presenta una progressività nel corso degli anni (Kumar et al.. 1984).
La funzionalità vestibolare è caratterizzata da areflessia nella sindrome di Usher Tipo I ,da
normoreflessia nella sindrome di Usher Tipo II e da risposte che vanno progressivamente peggiorando nella sindrome di Usher Tipo III (Kumar et al. 1984: Möller et al.. 1989)
|
Tipo |
Ipoacusia |
Funzionalità Vestibolare |
Comparsa della retinite pigmentosa |
|
Tipo I |
Profonda Congenita |
Compromessa |
Durante la prima decade |
|
Tipo II |
Audiogramma in discesa Congenita |
Normale |
Durante la prima o la seconda decade |
|
Tipo III |
Progressiva |
Variabile |
Variabile |
Tab. III
La maggior parte dei soggetti affetti da sindrome di Usher presentano un normale sviluppo cognitivo e neurologico.
La diagnosi precoce è molto importante al fine del programma terapeutico-riabilitativo. La prognosi infatti è caratterizzata dalla comparsa di una cecità attorno ai 3 0-40 anni di età che andrà a sommarsi al deficit uditivo. compromettendo notevolmente il grado di autonomia del soggetto
1.10.6.1 b)la sindrome di Alport
La sindrome di Alport rappresenta la forma più frequente tra le sindrome associate a anomalie renali, È responsabile di circa l’1% delle ipoacusie neurosensoriali congenite.
La sindrome di Alport è causata da una eterogeneità di mutazione che si esprimono soprattutto a carico dei geni COL4A3. COL1A1 o COL4A5 che codificano per il collagene di tipo IV La forma più frequente. caratterizzata da mutazione nel gene COL1A5 localizzato nella regione Xq22. è caratterizzata da una modalità di trasmissione X-linked dominante /Flinter et al.. 1990). Il gene COL4A5 coinvolto codifica per la catena 5 del collagene tipo IV ed è stato localizzato nella coclea a livello della membrana basilare. del ligamento spirale e della stria vascularis (Atkin et aL.1988 Circa l’85% dei casi di sindrome di Alport presenta pattern di trasmissione X-linked. mentre circa il 150/o presenta altrc modalità trasmissive. Le forme più rare sono legate a mutazione dei geni COL1A3 e COL4A1. con locus 2q36-q37 e presentano modalità trasmissiva autosomica recessiva (Gubler et al.. 1995. Lemmink, 1994).
Nel 1927 Alport descrisse per la prima volta una famiglia i cui membri presentavano sordità associata a disfunzione renale, in cui i maschi morivano per uricemia. per questo motivo il suo nome fu utilizzato per descrivere una nefropatia familiare progressiva che si manifesta con una ematuria associata ad ipoacusia. particolarmente grave nei maschi.
La sintomatologia a carico del sistema renale è simile nelle varie nelle varie forme. La forma X-linked può essere caratterizzata da insorgenza precoce che rende necessaria la dialisi entro i 20 anni e conduce alla morte entro il 31° anno di vita oppure da insorgenza tardiva con necessità di dialisi dopo i 40 anni. Questa variabilità è probabilmente legata al tipo di mutazione genetica. L’ematuria è il segno clinico principale ed è presente nei maschi con frequenza doppia rispetto alle femmine. Proteinuria e albuminuria sono segni meno frequenti nell’infanzia che tendono a peggiorare negli anni (Grunfeld, 1985).
Circa il 55% dei maschi affetti e il 40% delle femmine presentano un’ipoacusia neurosensoriale accentuata sulle frequenze acute di tipo progressivo. Solitamente l’ipoacusia si istaura durante la seconda decade e nelle femmine insorge più tardivamente (Gleeson, 1984). A carico dell’occhio circa il 35-70% dei soggetti con forma X-linked ad insorgenza giovanile presenta cheratocono e sviluppa una miopia assiale (Rhys et al. 1997).
Al fine della diagnosi è necessaria la presenza di almeno tre dei seguenti criteri;
1) familiarità positiva per ematuria +/- insufficienza renale:
2) biopsia renale con aspetti tipici alla microscopia elettronica:
3) segni oftalmologici caratteristici:
4) ipoacusia neurosensoriale per le frequenze acute progressiva durante l’infanzia.
La prognosi è variabile: per alcuni soggetti i disturbi possono essere di grado lieve. mentre per altri può essere necessaria la dialisi e il successivo trapianto renale già in età precoce Jais et al.. 2000).
BIBLIOGRAFIA
Abdelhak S., Kalatzis V., Heilig R., Compain S., Samson D., Vincent C., e al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family Nat. Genet. 1997 ; 15 : 157-164 [cross-ref]
Albert S., Blons H., Denoyelle F., Feldmann D., Loundon N., Sergent-Allaoui A., e al. LC26A4 gene is frequently involved in non syndromic hearing impairment with inner ear malformation in Caucasian populations J. Med. Genet. 2005 ; (soumis).
Amiel J., Attiee-Bitach T., Marianowski R., Cormier-Daire V., Abadie V., Bonnet D., e al. Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome Am. J. Med. Genet. 2001 ; 99 : 124-127 [cross-ref]
Andreas R, Janecke AR, Hirst-Stadlmann A, Gunther B, Utermann B, Müller T. et al Progressive hearing loss and recurrent sudden sensoryneural hearing loss associated with GJB2 mutations phenotypic spectrum and frequencies of GJB2 mutations in Austria Hum. Genet 2002: 111:115-153
App SA. Rankin WA. Kurmis AP Connexin 26 mutations in autosomal recessive deafness disorders:. a review . Int. J. Audiol 2007 Feb:46( 2):.75-81
Armitage I.M., Burke J.P., Buffin J.T. Visual impairment in severe and profound sensorineural deafness Arch. Dis. Child. 1995 ; 73 : 53-56 [cross-ref]
Baker KA. Dhyana S. Chien E . Rua Y. Yeager M. Stevens RC Expression Screening and Thermal Stability of Human Connexin 26 Protein Expr. Purif. (2007) 56(1):85-92.
Batissoco AC, Abreu-Silva RS, Braga MC, Lezirovitz K, Della-Rosa V, Alfredo T Jr, Otto PA. Mingroni-Netto RC, Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: implications for diagnosis and genetic counselling Ear Rear. 2009 Feb:30(1): 1-7
Bernardes R. Bortoncello S. Christiani TV. Sartorato EL. Silva RC. Porto PR Molecular investigation in children candidates and submitted to cochlear implantation Rev Bras Otorrinolaringol ,2006:72(3):333-6
Bespalova I.N., Van Camp G., Bom S.J., Brown D.J., Cryns K., DeWan A.T., e al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss Hum. Mol. Genet. 2001 ; 10 : 2501-2508 [cross-ref]
Bicego M, Beltramello M, Melchionda S, Carella M, Piazza V, Zelante L et al. Pathogenetic role of the deafness-related M34T mutation ofCx26. Hum Mol Genet. 2006 Sep 1: 15(17):2569-87.
Bilgin H., Kasemsuwan L., Schachern P.A., Paparella M.M., Le C.T. Temporal bone study of Down’s syndrome Arch. Otolaryngol. Head Neck Surg. 1996 ; 122 : 271-275
Blons H., Feldmann D., Duval V., Messaz O., Denoyelle F., Loundon N., e al. Screening of SLC26A4 (PDS) gene in Pendred’s syndrome: a large spectrum of mutations in France and phenotypic heterogeneity Clin. Genet. 2004 ; 66 : 333-340 [cross-ref]
Boothroyd A. & Cawkwell S. Vibrotactile thresholds in pure tone audiometry Acta Otolaryngologica. 1970: 69.381-387
Brobby G.W., Muller-Myhsok B., Horstmann R.D. Connexin 26 R143W mutation associated with recessive non syndromic sensorineural deafness in Africa N. Engl. J. Med. 1998 ; 338 : 548-550
Cama E, Melchionda S, Palladino T, Carella M, Santarelli R, Genovese E, Benettazzo F, Zelante L, Arslan E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort Int. J. Audiol 2009 Jan:48(1): 12-7
Campbell C., Cucci R.A., Prasad S., Green G.E., Edeal J.B., Galer C.E., e al. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations Hum. Mutat. 2001 ; 17 : 403-411 [cross-ref]
Carrasquillo MM, Zlotogora I, Barges S, Chakravarti A. Two different connexin 26 mutations in an inbred kindred segregating non-syndromic recessive deafness. implications for genetic studies in isolated populations Hum. Molec. Genet. 6. 2163-2172. 1997.
Chen Q., Zhang D., Gingell R.L., Moss A.J., Napolitano C., Priori S.G., e al. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome Circulation 1999 ; 99 : 1344-1347
COMITATO NAZIONALE PER LA BIOSICUREZZA E LE BIOTECNOLOGIE – ISS. Linee guida per test genetici. Rapporto del Gruppo di lavoro 1998
Coucke P.J., Van Hauwe P., Kelley P.M., Kunst H., Schatteman I., Van Velzen D., e al. Mutations in the KCNQ4 gene are responsible for autosomal dominant deafness in four DFNA2 families Hum. Mol. Genet. 1999 ; 8 : 1321-1328 [cross-ref]
Cremers F.P., Cremers C.W., Ropers H.H. The ins and outs of X-linked deafness type 3 Adv. Otorhinolaryngol. 2000 ; 56 : 184-195 [cross-ref]
Cohen-Salmon M., Ott T., Michel V., Hardelin J.P., Perfettini I., Eybalin M., e al. Targeted ablation of connexin 26 in the inner ear epithelial gap junction network causes hearing impairment and cell death Curr. Biol. 2002 ; 12 : 1106-1111 [cross-ref]
Coyle B., Coffey R., Armour J.A., Gausden E., Hochberg Z., Grossman A., e al. Pendred syndrome (goitre and sensorineural hearing loss) maps to chromosome 7 in the region containing the non syndromic deafness gene DFNB4 Nat. Genet. 1996 ; 12 : 421-423 [cross-ref]
Coyle B., Reardon W., Herbrick J.A., Tsui L.C., Gausden E., Lee J., e al. Molecular analysis of the PDS gene in Pendred syndrome (sensorineural hearing loss and goitre) Hum. Mol. Genet. 1998 ; 7 : 1105-1112 [cross-ref]
Cryns K. Orzan E. Murgia A. Huygen PLM. Moreno F. del Castillo I. Parker Chamberlin G. Azaiez H. Prasad S, Cucci RA. Leonardi E. Snoeckx RL. Govaerts PJ. Van de Heyning PH. Van de Heyning CM. Srnith RJH. Van Camp G. A genotype-phenotype correlation for G.JB2 (connexin 26) deafness J. Med. Genet. 2004 ; 41 : 147-154 [cross-ref]
D’Andrea P. Veronesi V. Bicego M. Melchionda 5. Zelante L. Di brio E. Bruzzone R. Gasparini P. Rea- ring bss. frequencv and functional studies of the most common connexin26 alleles. Biochemical and Biophysical Research Communications 296 (2002) 685-69 1
Dahl R-H M, Tobin SE. Poulakis Z. Rickards FW. Xu X. Gillam L. et al The contribution of GJB2 mutations to slight or mild hearing bss in Australian elementary school children. J Med Genet 2006:43:850-855.
de Kok Y.J., van der Maarel S.M., Bitner-Glindzicz M., Huber I., Monaco A.P., Malcolm S., e al. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4 Science 1995 ; 267 : 685-688
del Castillo I., Villamar M., Moreno-Pelayo M.A., del Castillo F.J., Alvarez A., Telleria D., e al. A deletion involving the connexin 30 gene in non syndromic hearing impairment N. Engl. J. Med. 2002 ; 346 : 243-249 [cross-ref]
Denoyelle F., Weil D., Maw M.A., Wilcox S.A., Lench N.J., Allen-Powell D.R., e al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin26 gene Hum. Mol. Genet. 1997 ; 6 : 2173-2177 [cross-ref]
Denoyelle F., Lina-Granade G., Plauchu H., Bruzzone R., Chaib H., Levi-Acobas F., e al. Connexin26 gene linked to a dominant deafness Nature 1998 ; 393 : 319-320 [cross-ref]
Denoyelle F., Petit C., Garabedian E.N., Marlin S. Diagnostic étiologique des surdités de perception de l’enfant: bilan d’un an de consultation de conseil génétique des surdités Ann. Otolaryngol. Chir. Cervicofac. 1998 ; 115 : 3-8
Denoyelle F., Marlin S., Weil D., Moatti L., Chauvin P., Garabédian E.N., e al. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin 26 gene defect: implications for genetic counselling Lancet 1999 ; 353 : 1298-1303 [cross-ref]
Denoyelle F et al “GIB2 and GIB6 Mutations Genotypic and Phenotypic Correlations in a Large Cohort of Rearing-Irnpaired Patients Arch Otolaringol Head Neck Surgery 2005: 131:481:487
El-Schahawi M., deMunain L., Sarrazin A.M., Shanske A.L., Basirico M., Shanske S., e al. Two large Spanish pedigrees with non-syndromic sensorineural deafness and the mtDNA mutation at nt 1555 in the 12SrRNA gene: evidence of heteroplasmy Neurology 1997 ; 48 : 453-456
Erbe CB. Harris KC. Runge-Samuelson CL. Flanary VA. Wackym PA Connexin 26 and connexin 30 rnutations in children with non syndromic hearing bss Laryngoscope 2004 Apr:114(4):607-l1
Estivill X., Govea N., Barcelo E., Badenas C., Romero E., Moral L., e al. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides Am. J. Hum. Genet. 1998 ; 62 : 27-35 [cross-ref]
Everett L.A., Glaser B., Beck J.C., Idol J.R., Buchs A., Heyman M., e al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat. Genet. 1997 ; 17 : 411-422 [cross-ref]
Evirgen N. Solak M. Derekòy 5. Erdoan M. Yildiz R. Eser B. Arikan S.. Erko A Genotyping for Cx26 and Cx30 nnitations in cases with congenital hearing loss. Genetic Testing. lune 1. 2008. 12(2): 253-256.
Feldmann D., Denoyelle F., Loundon N., Weil D., Garabedian E.N., Couderc R., e al. Clinical evidence of the nonpathogenic nature of the M34T variant in the connexin 26 gene Eur. J. Hum. Genet. 2004 ; 12 : 279-284 [cross-ref]
Fichel-Ghodsian N. Mitochondrial deafness Ear Hear. 2003 ; 24 : 303-313
Fraser F.C., Sproule J.R., Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss Am. J. Med. Genet. 1980 ; 7 : 341-349 [cross-ref]
Friedman R.A., Bykhovskaya Y., Tu G., Talbot J.M., Wilson D.F., Parnes L.S., e al. Molecular analysis of the POU3F4 gene in patients with clinical and radiographic evidence of X-linked mixed deafness with perilymphatic gusher Ann Otol Rhinol Laryngol 1997 ; 106 : 320-325
Fuse Y., Doi K., Hasegawa T., Sugii A., Hibino H., Kubo T. Three novel connexin 26 gene mutations in autosomal recessive non-syndromic deafness Neuroreport 1999 ; 10 : 1853-1857
Gasparini P., Rabionet R., Barbujani G., Melchionda S., Petersen M., Brondum-Nielsen K., e al. High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG Eur. J. Hum. Genet. 2000 ; 8 : 19-23 [cross-ref]
Genetic Evaluation of Congenital Hearing Loss Expert Panel Genetics Evaluation Guidelines for the Etiologic Diagnosis of Congenital Hearing Loss Genet Med 2002:1(3). 162-171
Gorlin R.J., Toriello H.V., Cohen M.M. Hereditary hearing loss and its syndromes New York: Oxford University Press (1995).
Green GE. Mueller RE. Cohn ES.Avraham KB. Kanaan MB. Smith RJH Audiological manifestations and features of connexin 26 deafness Audiological Medicine 2003: 1.5-11
Gualandi E. Ravani A. Berto A. Burdo S. Trevisi P. Ferlini A. Martini A. Calzolari E Occurrence of Del(GJB6 – D13S1830) Mutation in Italian Non-syndromic Hearing Loss Patients Carryng a Single GJB2 Mutated Allele Acta OtoIaryngol 2004: Suppl 552:29-34
Gualandi E. Ravani A. Berto A. Sensi A. Trabanelli C. Falciano E.et al. Exploring the clinical and epidemiological complexity of GJB2—linked deafness Am J Med Genet 2002 Sep 15:112(1)38-45
Hall B.D. Choanal atresia and associated multiples anomalies J. Pediatr. 1979 ; 95 : 395-398 [cross-ref]
Hanson MA. Brooun A. Baker KA .Jaakola VP. Roth C. Chien EY. et al Profiling of membrane protein variants in a haculovirus system by coupling cell-surface detection with smalI-scale paralleI expression Protein Expr Purif. 2007: 56( I):.85-92
Hildesheimer M., Maayan Z., Muchnik C., Rubinstein M., Goodman R.M. Auditory and vestibular findings in Waardenburg’s type II syndrome J. Laryngol. Otol. 1989 ; 103 : 1130-1133
Hwa HL. Ko TM. Hsu CJ.Chiang YL. Oong JL. Chen CC. et al. Mutation spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with prelingual deafness Genet Med 2003: 5:161-165
Jais J.P., Knebelmann B., Giatras I., De Marchi M., Rizzoni G., Renieri A., e al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study J. Am. Soc. Nephrol. 2003 ; 14 : 2603-2610 [cross-ref]
Kelley PM. Harris Di. Comer BC, Askew JW. Fowier T. Smith SD. et al Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB 1) hearing loss. Am i Hum Genet 1998; 62:792—799
Kelsell D.P., Wilgoss A.L., Richard G., Stevens H.P., Munro C.S., Leigh I.M. Connexin mutations associated with palmoplantar keratoderma and profound deafness in a single family Eur. J. Hum. Genet. 2000 ; 8 : 469-472
Kelsell D.P., Dunlop J., Stevens H.P., Lench N.J., Liang J.N., Parry G., e al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness Nature 1997 ; 387 : 80-83 [cross-ref]
Kenneson A. Van Naarden Braun K. Boyle C. GJB2 (connexin 26) variants and non syndromic sensorineural hearing bss. A HuGE review. Genet Med 2002 Jul-Aug:4(4):258-74
Khanim F., Kirk J., Latif F., Barrett T.G. WFS1/Wolfram in mutations, Wolfram syndrome, and associated diseases Hum. Mutat. 2001 ; 17 : 357-367 [cross-ref]
Kubisch C., Schroeder B.C., Friedrich T., Lutjohann B., El-Amraoui A., Marlin S., e al. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness Cell 1999 ; 96 : 437-446 [cross-ref]
Kudo T., Ikeda K., Kure S., Matsubara Y., Oshima T., Watanabe K., e al. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population Am. J. Med. Genet. 2000 ; 90 : 141-145 [cross-ref]
Kumar NM. Gilula NB. et al The Gap Junction Review Communication Channel. Cell 1996: VoI. 84,381—388.
Lai-Cheong JE. Arita K. McGrath .1 Genetic Disease of Junctions. .Journal of lnvestigative Dermatology 2007: 127,2713-2725
Lerer I., Sagi M., Ben-Neriah Z., Wang T., Levi H., Abeliovich D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews Hum. Mutat. 2001 ; 18 : 460 [cross-ref]
Li X.C., Everett L.A., Lalwani A.K., Desmukh D., Friedman R.B., Green E.D., e al. A mutation in PDS causes non-syndromic recessive deafness Nat. Genet. 1998 ; 18 : 215-217 [cross-ref]
Liu X.Z., Xia X.J., Adams J., Chen Z.Y., Welch K.O., Tekin M., e al. Mutations in GJA1 (connexin 43) are associated with non-syndromic autosomal recessive deafness Hum. Mol. Genet. 2001 ; 10 : 2945-2951 [cross-ref]
Liu X.Z., Ouyang X.M., Xia X.J., Zheng J., Pandya A., Li F., e al. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss Hum. Mol. Genet. 2003 ; 12 : 1155-1162 [cross-ref]
Liu XZ. Pandya A .Angeli S . Tebischi FF. Arnos KS, Nance WE. Baikany T Audiological Features of GJB2 (Connexin 26) Deafness Ear & Hearing 2005: 26: 361-369.
Loundon N , Marcolla A , Roux io , Rouillon io , Denoyelle F , Feldmann D , Marlin S , Garabedian EN. Auditory neuropathy or endocochlear hearing loss? Otol Neurotol 2005 ; 26 (4) :748-54.
Madden C., Rutter M., Hilbert L., Greinwald J.H., Choo D.I. Clinical and audiological features in auditory neuropathy Arch. Otolaryngol. Head Neck Surg. 2002 ; 128 : 1026-1030
Maestrini E., Korge B.P., Ocana-Sierra J., Calzolari E., Cambiaghi S., Scudder P.M., e al. Missense mutation in connexin26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel’s syndrome) in three unrelated families Hum. Mol. Genet. 1999 ; 8 : 1237-1243 [cross-ref]
Marlin S. Surdités génétiques Génétique médicale Paris: Masson (2004). 281-299
Marlin S., Feldmann D., Blons H., Loundon N., Rouillon I., Albert S., e al. GJB2 and GJB6 mutations: genotypic and phenotypic correlation in a large cohort of hearing-impaired patients Arch. Otolaryngol. Head Neck Surg. 2005 ; (in press).
Maroudias N., Economides J., Christodoulou P., Helidonis E. A study on the otoscopical and audiological findings in patients with Down’s syndrom in Greece Int J Pediatr Otolaryngol 1994 ; 29 : 143-149
Mehl AL. Thornson V The Colorado newborn hearing screening project. 1992-1999. on the threshold of effective population-based universal newborn hearing screening. Pediatrics 2002: 109:E7
Mese G. Londin E. Mui R. Brink PR. White TW. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing bss, Hum. Genet. 115. 191-199. 2004
Migliosi V., Modamio-Hoybjor S., Moreno-Pelayo M.A., Rodrigues-Ballesteros M., Villamar M., Telleria D., e al. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss J. Med. Genet. 2002 ; 39 : 502-506 [cross-ref]
Morell R.J., Kim H.J., Hood L.J., Goforth L., Friderici K., Fisher R., e al. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with non syndromic recessive deafness N. Engl. J. Med. 1998 ; 339 : 1500-1505 [cross-ref]
Morlé L., Bozon M., Alloisio N., Latour P., Vandenberghe A., Plauchu H., e al. A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss J. Med. Genet. 2000 ; 37 : 368-369
Morton N.E. Genetic epidemiology of hearing impairment. in Genetics of hearing impairment Ann. N. Y. Acad. Sci. 1991 ; 630 : 16-31 [cross-ref]
Neyroud N., Richard P., Vignier N., Donger C., Denjoy I., Demay L., e al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome Circ. Res. 1999 ; 84 : 290-297
Nickel R. Forge A Gap junctions and connexins in the inner ear: their roles in homeostasis and deafness. Curr Opin Otolaryngol Head Neck Surg 2008 Oct:16(5):452-7
Ouyang X. Mutational analysis of the six cloned Usher genes using a sequential screening strategy Florida: Communication, 26th ARO meeting (2000).
Pabla H.S., McCormick B., Gibbin K.P. Retrospective study of the prevalence of bilateral sensorineural deafness in childhood Int. J. Pediatr. Otorhinolaryngol. 1991 ; 22 : 161-165 [cross-ref]
Pallares-Ruiz N., Blanchet P., Mondain M., Claustres M., Roux A.F. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur. J. Hum. Genet. 2002 ; 10 : 72-76 [cross-ref]
Park Hi. Hahn SH. Chun YM. Park K. Kim EN Connexin 26 mutations associated with nonsyndromic hearing bss Laryngoscope 2000; 110.9. 1535-1538.
Parving A. Hearing disorders in childhood, some procedures for detection, identification and diagnostic evaluation Int. J. Pediatr. Otorhinolaryngol. 1985 ; 9 : 31-57
Paznekas W.A., Boyadjiev S.A., Shapiro R.E., Daniels O., Wollnik B., Keegan C.E., e al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia Am. J. Hum. Genet. 2003 ; 72 : 408-418 [cross-ref]
Petersen MB. Wiliems P.J Non-syndromic. autosomal-recessive deafness. Clin Genet 2006 Mav:695 37 1-92
Petit C., Levilliers J., Marlin S., Hardelin J.P. Hereditary hearing loss Metabolic and molecular basis of inherited disease New York: McGraw-Hill Publishers (2001). 6281-6328
Phelps P.D., Coffey R.A., Trembath R., Luxon L., Grossman A., Britton K.E., e al. Radiological malformations of the ear in Pendred syndrome Clin. Radiol. 1998 ; 53 : 268-273 [cross-ref]
Piatto VB. Moreira OA. Silva MA. Maniglia IV. Pereira MC. Sartorato EL Correlation between audiometric data an the 35delG mutation in ten patients. Rev Bras Otorrinolaringol 2007:73(6):777-83
Prezant T.R., Agapian J.V., Bohlman M.C., Bu X., Oztas S., Qiu W.Q., e al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness Nat. Genet. 1993 ; 4 : 289-294 [cross-ref]
Roger G., Morisseau-Durand M.P., Van den Abbeele T., Nicollas R., Triglia J.M., Narcy P., e al. The CHARGE association: the role of tracheotomy Arch. Otolaryngol. Head Neck Surg. 1999 ; 125 : 33-38
Richard G., Rouan F., Willoughby C.E., Brown N., Chung P., Ryynanen M., e al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome Am. J. Hum. Genet. 2002 ; 70 : 1341-1348 [cross-ref]
Robertson N.G., Skvorak A.B., Yin Y., Weremowicz S., Johnson K.R., Kovatch K.A., e al. Mapping and characterization of a novel cochlear gene in human and in mouse: a positional candidate gene for a deafness disorder, DFNA9 Genomics 1997 ; 46 : 345-354 [cross-ref]
Roux A.F., Pallares-Ruiz N., Vielle A., Faugere V., Templin C., Leprevost D., e al. Molecular epidemiology of DFNB1 deafness in France BMC Med. Genet. 2004 ; 5 : 1-10 [cross-ref]
Ruf R.G., Xu P.X., Silvius D., Otto E.A., Beekmann F., Muerb U.T., e al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes Proc. Natl. Acad. Sci. USA 2004 ; 101 : 8090-8095 [cross-ref]
Satar B., Mukherji S.K., Telian S.A. Congenital aplasia of the semicircular canals Otol. Neurotol. 2003 ; 24 : 437-446 [cross-ref]
Sheffield V.C., Kraiem Z., Beck J.C., Nishimura D., Stone E.M., Salameh M., e al. Pendred syndrome maps to chromosome 7q21-34 and is caused by an intrinsic defect in thyroid iodine organification Nat. Genet. 1996 ; 12 : 424-426 [cross-ref]
Schwarts D.M., Schwarts R.H. Acoustic impedance in young children with Down’s syndrome Arch. Otolaryngol. 1978 ; 107 : 652-656
Snoeckx RL. Hu gen PLM. Feidmann D. Marlin 5. Denoyeile F. Wahgora i. Mueiler-Maiesinska M. Agneszka Pollak A. Pioski R. Murgia A. Orzan E GJB2 Mutations and Degree of Hearing Loss. A Multicenter Study Am. .1. Hum Genet 77,935—957. 2005
Sobe I. VrciccIe S, Shahin H. Berlin M. Davis N. Kanaan M. et ai The prevalence and expression of inherited connexin 26 mutations associated with nonsyndromic hearing loss in the Israeli population Human Genetics 2000: 106.50-57
Splawski I., Timothy K.W., Vincent G.M., Atkinson D.L., Keating M.T. Molecular basis of the long-QT syndrome associated with deafness N. Engl. J. Med. 1997 ; 336 : 1562-1567 [cross-ref]
Starr A., Picton T.W., Sihinger Y., Hood L.J., Berlin C.I. Auditory neuropathy Brain 1996 ; 119 : 741-753 [cross-ref]
Stojkovic T., Latour P., Vandenberghe A., Hurtevent J.F., Vermersch P. Sensorineural deafness in X-linked Charcot-Marie-Tooth disease with connexin 32 mutation (R142Q) Neurology 1999 ; 52 : 1010-1014
Talbot J.M., Wilson D.F. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapes footplate and perilymphatic gusher Am. J. Otol. 1994 ; 15 : 177-182
Tassabehji M., Newton V.E., Liu X.Z., Brady A., Donnai D., Krajewska-Walasek M., e al. The mutational spectrum in Waardenburg syndrome Hum. Mol. Genet. 1995 ; 4 : 2131-2137
Tekin M, Duman T, Boğoçlu G, Incesulu A, Comak E, Ilhan I, Akar N. Moderate hearing loss and pseudo dominant inheritance due to L9OP/35deiG mutations in the GJB2 (connexin 26) gene. Genet Couns 2003: 14(4),37986
Tranebaerg L Genetics of congenital hearing impairment: a clinical approach mt i Audiol 2008 Sep:47(c. :535-45
Welch KQ. NI;crjn RS. Pandya A. Amos KS. Compound heterozygosity for dominant and recessive GJB2 ncutations, Effect on phenotype and review of the literature. Am J Med Genet Part A 143A 1567-1573 2007.
Usami S., Abe S., Weston M.D., Shinkawa H., Van Camp G., Kimberling W.J. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations Hum. Genet. 1999 ; 104 : 188-192 [cross-ref]
Van Camp G, Smith RJ. Hereditary hearing loss homepage, http://dnalab-www.uia.ac.be/dnalab/hhh.
Van Hauwe P., Everett L.A., Coucke P., Scott D.A., Kraft M.L., Ris-Stalpers C., e al. Two frequent missense mutations in Pendred syndrome Hum. Mol. Genet. 1998 ; 7 : 1099-1104 [cross-ref]
Vincent C., Kalatzis V., Abdelhak S., Chaib H., Compain S., Helias J., e al. BOR and BO syndromes are allelic defects of EYA1 Eur. J. Hum. Genet. 1997 ; 5 : 242-246
Verhagen W.I., Bom S.J., Fransen E., Van Camp G., Huygen P.L., Theunissen E.J., e al. Hereditary cochleovestibular dysfunction due to a COCH gene mutation (DFNA9): a follow-up study of a family Clin. Otolaryngol. 2001 ; 26 : 477-483 [cross-ref]
Vissers L.E., Van Ravenswaaij C.M., Admiraal R., Hurst J.A., De Vries B.B., Janssen I.M., e al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome Nat. Genet. 2004 ; 36 : 955-957 [cross-ref]
Wang Z., Li H., Moss A.J., Robinson J., Zareba W., Knilans T., e al. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome Mol. Genet. Metab. 2002 ; 75 : 308-316 [cross-ref]
Wiliems PJ Genetic causes of hearing bss New Eng. J Med 332: 1101-1109. 2000
Yum SW. Zhang J. Valiunas V. Kanaporis G. Brink PR. White 1W. Seherer SS. Human connexin26 and C000C\itt3() form functional heteromeric and heterotypic channels. Am .1 Physiol Cell Physiol. 2007 Sep: 293 3:C1032-4S
Yasunaga S., Grati M., Cohen-Salmon M., El-Amraoui A., Mustapha M., Salem N., e al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a non syndromic form of deafness Nat. Genet. 1999 ; 21 : 363-369
Yoshida M., Shintani T., Hirao M., Himi T., Yamaguchi A., Kikuchi K. Aminoglycoside-induced hearing loss in a patient with the 961 mutation in mitochondrial DNA ORL J. Otorhinolaryngol. Relat. Spec. 2002 ; 64 : 219-222 [cross-ref]
Young T.L., Ives E., Lynch E., Person R., Snook S., MacLaren L., e al. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1 Hum. Mol. Genet. 2001 ; 10 : 2509-2514 [cross-ref]
Zelante L., Gasparini P., Estivill X., Melchionda S., D’Agruma L., Govea N., e al. Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans Hum. Mol. Genet. 1997 ; 6 : 1605-1609 [cross-ref]
Zhao H., Li R., Wang Q., Yan Q., Deng J.H., Han D., e al. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family Am. J. Hum. Genet. 2004 ; 74 : 139-152 [cross-ref]
http: ghr nlm
OMIM
The Connexin deafness home page
http//: hereditary hearing loss, org