Geni della connexina
SORDITA ‘, AUTOSOMICA 1A RECESSIVA; DFNB1A
|
|
||||||||||||||||||||||||||||
|
SORDITA ‘, DIGENICA, GJB2 / GJB6, INCLUSO |
||||||||||||||||||||||||||||
|
SORDITA ‘, DIGENICA, GJB2 / GJB3, INCLUSO |
||||||||||||||||||||||||||||
|
Rapporti fenotipo-Gene |
||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||
Qual è il nome ufficiale del gene GJB2 ?
Il nome ufficiale di questo gene è “gap junction proteina beta 2.”
GJB2 è il simbolo ufficiale del gene. Il GJB2 gene è conosciuto anche con altri nomi, elencati di seguito.
Per saperne di più sui nomi geni e simboli sulla About pagina.
Qual è la funzione normale del gene GJB2 ?
Il GJB2 gene fornisce istruzioni per fare una proteina chiamata gap junction beta 2, più comunemente noto come connessina 26. Connexin 26 è un membro della famiglia di proteine connessina. Proteine Connexin formano canali chiamati giunzioni che consentono il trasporto di sostanze nutritive, pagano atomi (ioni), e molecole di segnalazione tra cellule che sono in contatto tra loro vicini. La dimensione della giunzione gap ed i tipi di particelle che si muovono attraverso di esso sono determinate dalle particolari proteine connessine che compongono il canale. Giunzioni realizzate con connessina 26 ioni di potassio di trasporto e di alcune piccole molecole.
Connexin 26 si trova nelle cellule in tutto il corpo, compreso l’orecchio interno e la pelle. A causa della sua presenza nell’orecchio interno, in particolare la struttura a forma di chiocciola chiamata coclea, i ricercatori sono interessati a ruolo di questa proteina in udienza. Hearing implica la conversione delle onde sonore ad impulsi nervosi elettrici. Questa conversione coinvolge molti processi, tra cui il mantenimento del corretto livello di ioni potassio nell’orecchio interno. Alcuni studi indicano che i canali realizzati con connessina 26 aiuto a mantenere il corretto livello di ioni di potassio. Altre ricerche indicano che connessina 26 è necessario per la maturazione di alcune cellule nella coclea. Connexin 26 gioca anche un ruolo nella crescita, la maturazione e la stabilità dello strato più esterno della pelle (epidermide).
Le connessine sono una famiglia di proteine di membrana largamente espresse nell’organismo umano (finora ne sono stati identificati più 20 tipi diversi); ogni connessina è costituita da quattro domini transmembrana con due loops extracellulari ed uno citoplasmatico e si combina in esameri formare un complesso che viene definito connessone (fig. 1); ogni connessone, a sua volta, interagisce con un altro connessone di una cellula adiacente per formare canali intercellulari o gap junctions: tali strutture sono molto importanti per gli scambi di elettroliti, secondi messaggeri e metaboliti 3,9.
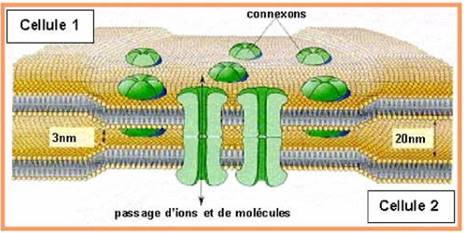
Fig. 1. Struttura di una gap junction
Grazie a studi effettuati nell’epitelio sensoriale dell’orecchio interno di rettili, volatili e mammiferi è stato possibile accertare e individuare i diversi tipi di connessina che costituiscono le “gap junctions” delle cellule sensoriali dell’orecchio interno dei mammiferi. Da questi studi è emerso che nella coclea dei mammiferi sono presenti quattro isotipi (cx 26, cx30, cx31, cx43) mentre nell’organo vestibolare ne sono presenti tre (cx26, cx30, cx43). La cx26 e la cx30 sono state trovate nella regione basale della stria vascolare, nelle cellule del lembo spirale e nelle cellule di sostegno dell’organo del Corti (Fig. 2).
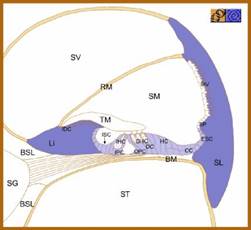
![]() Fig. 2- Sezione della coclea: in viola zone di espressione di cx26 e cx30
Fig. 2- Sezione della coclea: in viola zone di espressione di cx26 e cx30
La cx31 è collocata nelle cellule situate a livello della prominenza spirale, una regione dove la cx30 non è espressa e la percentuale di cx26 è molto bassa (Fig. 3).
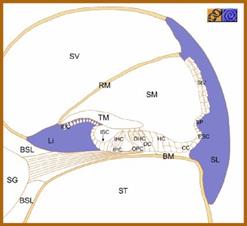
Figura 3- Sezione della coclea: in viola zone di espressione di cx31
La cx43 è stata individuata solo in alcuni tipi di fibrociti (10).
La mutazione più comune nella connessina 26 è la delezione in posizione 35 di una
Clinicamente l’ipoacusia associata alla mutazione 35delG è ad insorgenza preverbale, è neurosensoriale e non è progressiva (11).
Caratteristiche di condivisione fra i geni GJB2 con altri geni?
Il GJB2 gene appartiene ad una famiglia di geni chiamati GJ (proteine gap junction (connessine)).
Una famiglia genica è un gruppo di geni che condividono caratteristiche importanti. Classificare singoli geni in famiglie aiuta i ricercatori descrivono come i geni sono legati gli uni agli altri. Per ulteriori informazioni, vedere Quali sono famiglie di geni? Nel manuale.
Come sono cambiamenti nel gene GJB2 correlato alle condizioni di salute?
Sindrome di Bart-Pumphrey – causata da mutazioni nel GJB2 gene
Almeno due GJB2 mutazioni del gene sono state identificate le persone con sindrome di Bart-Pumphrey. Questa condizione è caratterizzata da una colorazione bianco dei chiodi (leuconichia), ispessite pelle sui palmi delle mani e piante dei piedi (cheratoderma palmo-plantare), escrescenze simili a verruche (pale nocche) sulle nocche delle dita delle mani e dei piedi, e la perdita dell’udito. I GJB2 mutazioni genetiche che causano la sindrome Bart-Pumphrey sostituire la glicina con la serina amino acido in posizione proteina 59 (Gly59Ser o G59S) proteina building block (amminoacido) o sostituire l’aminoacido asparagina con la lisina ammino acido in posizione proteina 54 ( Asn54Lys o N54K). La proteina alterata probabilmente interrompe la funzione di normale connessina 26 nelle cellule. Questa perturbazione potrebbe influenzare la crescita della pelle e anche mettere in pericolo l’udito disturbando la conversione delle onde sonore di impulsi nervosi.
ittiosi istrice-come con sordità – causata da mutazioni nel GJB2 gene
Almeno un GJB2 mutazione del gene è stato identificato nelle persone con ittiosi hystrix-come con sordità (HID), un disturbo caratterizzato da pelle secca e squamosa (ittiosi) e la perdita dell’udito che di solito è profonda. Questa mutazione sostituisce l’acido aspartico amminoacido con asparagina amino acido in posizione 50 della proteina, scritto come Asp50Asn o D50N.La mutazione è pensato di provocare canali che fuoriescono costantemente ioni, che compromette la salute delle cellule e aumenta la morte cellulare. La morte delle cellule della pelle e l’orecchio interno possono essere alla base dei segni e sintomi di HID.
Poiché la mutazione D50N può anche causare cheratite-ittiosi-sordità (KID) sindrome (descritto di seguito), molti ricercatori categorizzare sindrome KID e HID come una singola malattia, che loro chiamano KID / HID. Non è noto il motivo per cui alcune persone con questa mutazione hanno problemi agli occhi, mentre altri no.
Sindrome di cheratite-ittiosi-sordità – causata da mutazioni nel GJB2 gene
Almeno nove GJB2 mutazioni del gene sono state identificate in persone con cheratite-ittiosi-sordità (KID) sindrome, con il più comune è la mutazione D50N che si verifica anche in hystrix come ittiosi con sordità (sopra descritto). Sindrome KID è caratterizzata da cheratite, che è l’infiammazione della superficie anteriore dell’occhio (cornea); spesse chiazze arrossate di pelle secca e squamosa (ittiosi); e sordità.
I GJB2 mutazioni genetiche che causano la sindrome KID cambiamento singoli aminoacidi connessina 26. Le mutazioni si pensa al risultato nei canali che fuoriescono costantemente gli ioni, che danneggia la salute delle cellule e aumenta la morte cellulare. La morte delle cellule della pelle e l’orecchio interno può essere alla base della ittiosi e sordità che si verificano nella sindrome KID. Non è chiaro come GJB2 mutazioni genetiche colpiscono l’occhio.
Sordità sindromica – causata da mutazioni nel GJB2 gene
I ricercatori hanno identificato più di 90 GJB2 mutazioni genetiche che causano una forma di sordità non sindromica (perdita di udito senza segni e sintomi correlati che interessano altre parti del corpo) chiamato DFNB1. DFNB1 la sordità è ereditata come carattere autosomico recessivo, il che significa che due copie del GJB2 gene in ogni cellula sono alterati. GJB2mutazioni genetiche probabilmente alterano giunzioni, che possono disturbare il livello di ioni potassio nell’orecchio interno. Livelli di ioni potassio troppo elevati possono influenzare la funzione e la sopravvivenza delle cellule che sono necessarie per l’udito.
Alcune mutazioni eliminare o inserire elementi di DNA (coppie di basi) all’interno o nei pressi della GJB2 gene. La mutazione più comune, soprattutto nelle popolazioni bianche, elimina coppia di una base tra le posizioni 30 e 35 nel GJB2 gene (scritto come 35delG o 30delG).Nelle popolazioni asiatiche, una mutazione frequentemente riportati elimina una coppia di basi nella posizione 235 (235delC). Tra le persone con un orientale europea (Ashkenazi) antenati ebrei, una delezione di base-coppia alla posizione 167 (167delT) è una mutazione comune. Queste cancellazioni portano ad un anormalmente piccola proteina che non possono formare giunzioni funzionali.
Altri GJB2 mutazioni geniche sostituiscono uno degli aminoacidi utilizzati per rendere connessina 26 con un amminoacido non corretto. Queste mutazioni portano a una proteina instabile o deforme che non possono formare giunzioni, o una proteina alterata che forma giunzioni disfunzionali.
I ricercatori hanno anche identificato diversi GJB2 mutazioni genetiche che causano un’altra forma di sordità non sindromica chiamato DFNA3, che viene ereditato in modo autosomico dominante. Questo tipo di ereditarietà significa che una copia del GJB2 gene in ogni cella è alterata. Queste mutazioni sostituiscono un amminoacido in connessina 26 con un amminoacido non corretto. Non è ancora chiaro come questi GJB2 mutazioni genetiche portano alla perdita di udito DFNA3 associata. L’alterata connessina 26 proteina probabilmente inibisce il montaggio di giunzioni o la loro normale funzione, che potrebbero interrompere la conversione delle onde sonore di impulsi nervosi.
Cheratoderma palmoplantare con sordità – causata da mutazioni nel GJB2 gene
Almeno nove GJB2 mutazioni del gene sono state identificate nelle persone con cheratoderma palmoplantare con la sordità, una condizione caratterizzata da perdita e la pelle insolitamente spessa sentire sui palmi delle mani e piante dei piedi. I GJB2 mutazioni genetiche che causano cheratoderma palmo-plantare con sordità cambiano singoli aminoacidi in connessina 26. La proteina alterata probabilmente interrompe la funzione di normale connessina 26 nelle cellule, e possono interferire con la funzione di altre proteine connexin. Questa perturbazione potrebbe influenzare la crescita della pelle e anche mettere in pericolo l’udito disturbando la conversione delle onde sonore di impulsi nervosi.
Sindrome Vohwinkel – causata da mutazioni nel GJB2 gene
Almeno tre GJB2 mutazioni del gene sono state identificate le persone con sindrome di Vohwinkel. Questa condizione è caratterizzata da perdita dell’udito e la pelle anomalie. Oltre alle patch anormali di pelle, individui affetti sviluppano bande strette di tessuto fibroso anormale intorno le dita e dei piedi che può tagliare la circolazione alle cifre e provocare l’amputazione spontanea. I GJB2 mutazioni genetiche che causano la sindrome Vohwinkel cambiamento singoli aminoacidi in connessina 26. La proteina alterata probabilmente interrompe la funzione di normale connessina 26 nelle cellule, e può interferire con la funzione di altre proteine connessina. Questa perturbazione potrebbe influenzare la crescita della pelle e anche mettere in pericolo l’udito disturbando la conversione delle onde sonore di impulsi nervosi.
Dove si trova il GJB2 trova gene?
Citogenetica Località: 13q11-q12
Posizione molecolare sul cromosoma 13: coppie di basi 20.187.463 a 20,192,975
(Homo sapiens Annotazione di uscita 107, GRCh38.p2) (NCBI)![]()
![]()

Il GJB2 gene è situato sul lungo (q) braccio del cromosoma 13 tra le posizioni 11 e 12.
Più precisamente, il GJB2 gene si trova dalla coppia di basi 2
Citogenetica Località: 13q11-q12
Posizione molecolare sul cromosoma 13: coppie di basi 20.187.463 a 20,192,975
(Homo sapiens Annotazione di uscita 107, GRCh38.p2) (NCBI)![]()
Quadro clinico e terapia
Il quadro clinico si è detto esser caratterizzato da una sordità neurosensoriale bilaterale, di grado profondo, presente fin dalla nascita. Per questo tipo di deficit uditivo l’unica terapia è l’impianto cocleare. Questo dispositivo elettronico trasforma l’energia meccanica del suono in impulsi elettrici direttamente trasmessi al nervo acustico.
Analizzando la letteratura attualmente presente troviamo un numero esiguo di lavori; ciò è principalmente dovuto alla rarità della patologia. Cullen et al.[6] nel 2004 non hanno rilevato alcuna differenza dopo l’impianto nella percezione del parlato in open-set dei pazienti con mutazione rispetto
a quelli che presentavano sordità ma non associata a questa mutazione. Connell et al.[6] nel 2007 hanno evidenziato che sorprendentemente i pazienti con mutazione ottenevano migliori e più rapidi benefici nei test di linguaggio e comprensione, rispetto ai bambini con sordità neurosensoriali senza mutazione; questi infatti avevano alterazioni strutturali e molecolari più complesse. Questi risultati furono confermati anche da Bauer et al.[6] nel 2007 in uno studio di 4 bambini giapponesi omozigoti per GJB2; il livello di percezione del linguaggio post-impianto fu ritenuto migliore che in quelli che non presentavano la mutazione. Le considerazioni che si possono fare, tenendo conto di questi pareri discordanti, sono che non ci si attendono grandi risultati dall’impianto cocleare; la presenza della mutazione non è da considerarsi un indice prognostico negativo ma il margine di miglioramento atteso è sicuramente limitato.
1. Smith RJH, Hildebrand MS, Van Camp G et al. Deafnes and Hereditary Hearing Loss Overview. GeneReviews, Seattle 2010.
2. <<www.laboratoriogenoma.eu>>
3. Rossi G. Malattie dell’orecchio. In: Trattato di otorinolaringoiatria. Edizioni Minerva Medica, Torino 2004.
4. <<www.sciencephoto.com>>
5. <<www.sordita.it>>
Vivero RJ, Fan K, Angeli S et al. Cochlear implantation in common forms of genetic deafness. Int J Pediatr Otorhinolaryngol 2010;74(10):1107-1112
APPROFONDIMENTO
Un simbolo di cancelletto (#) e ‘di questa scheda l’evidenza che autosomica recessiva sordità-1A (DFNB1A) è causata da mutazione nel gene GJB2 (121011), che codifica il gap giunzione proteina connessina-26 (CX26), sul cromosoma . 13q11-q12, autosomica dominante sordità-3A (DFNA3A; 601.544) è una malattia allelica. Vedere anche DFNB1B (612.645), che è causata da una mutazione nel gene GJB6 (604.418) sul cromosoma 13q12.
Le connexine sono piccole proteine transmembranarie che si associano in complessi (connessoni) con funzione di canali di giunzione tra le cellule adiacenti per il trasporto di ioni implicati in particolare nei meccanismi di eccitazione delle cellule sensoriali cocleari. Attualmente nei mammiferi sono note cinque connexine diverse.
Nel 1997 è stato riconosciuto sul braccio lungo del cromosoma 13 il gene che codifica per la connessina 26 che è stato denominato GJB2 (Gap Junction Protein Beta 2) [1].
La connessina 26 è una proteina presente sulla membrana cellulare dove è coinvolta nella formazione delle gap junction, giunzioni che mettono in comunicazione cellule contigue permettendo il passaggio di piccole molecole e ioni. Nel 1997 è stato riconosciuto sul braccio lungo del cromosoma 13 il gene che codifica per la connessina 26 che è stato denominato GJB2 (Gap Junction Protein Beta 2) [1].
Dalla mutazione di origine ancora sconosciuta, di questo gene possono derivare 2 diverse forme di sordità ereditarie non sindromiche indicate con la sigla DFN, dall’inglese DeaFNess; quelle trasmesse con meccanismo autosomico dominante sono identificate dalla lettera A, quelle a trasmissione autosomica recessiva dalla lettera B. In modo specifico parliamo di DFNA3 per le autosomiche dominanti, che si manifesta in modo molto raro, mentre DFNB1 che costituisce circa l’80% dei casi di sordità genetica non sindromica autosomica recessiva in Italia e addirittura il 90% dei casi in Spagna. Quest’ultima mutazione è chiamata anche 35 del G, perchè comporta la perdita di una guanina in posizione 35. Il quadro clinico è caratterizzato da una sordità neurosensoriale bilaterale presente fin dalla nascita, di grado moderato o profondo, con andamento non progressivo [2].
Per effettuare diagnosi è utile l’analisi molecolare del gene in campioni biologici; il test può essere eseguito su prelievo ematico in EDTA (2 ml), liquido amniotico (10 ml), villi coriali (10 ml) e DNA (2 mcg) [2].
All’interno dell’organo dell’udito le gap junctions sono localizzate tra le cellule cigliate esterne e le cellule di sostegno. Il riciclo dello ione potassio endolinfatico durante la trasduzione sonora è giunzione-dipendente. L’espressione della connessina 26 nella coclea è indispensabile per la normale funzione dell’orecchio interno. La coclea è un piccolo condotto di 20-30 mm avvolto a spirale per 2 giri e 3/4. L’unione della lamina spirale ossea e della membrana basilare divide la coclea in due sezioni, una inferiore e una superiore. Quella inferiore è detta scala timpanica, quella superiore è scissa dalla membrana di Reissner in due compartimenti: uno superiore più ampio, detto scala vestibolare e uno inferiore più piccolo detto dotto cocleare (Figura 1). Nella scala vestibolare e nella scala timpanica vi è perilinfa che, come il liquido cefalorachidiano, ha una composizione simile ai liquidi extracellulari, con alta concentrazione di ioni sodio. Nel dotto cocleare vi è invece endolinfa che ha una maggior concentrazione di ioni potassio. Nel dotto cocleare sulla membrana basilare appoggia l’organo del Corti che attua la trasduzione dell’energia meccanica vibratoria in energia nervosa. L’organo del Corti è formato da cellule sensoriali sostenute dai pilastri di Corti interni ed esterni che circoscrivono una galleria triangolare. All’interno di essa si trovano circa 3400 cellule acustiche interne con aspetto globoso, mentre, all’esterno della galleria sono disposte su 3 file circa 30.000 cellule acustiche esterne di forma cilindrica allungata [3] (Figura 2 [4]). Pertanto le cellule acustiche si trovano all’interfaccia di due liquidi a composizione diversa. Le estremità apicali delle cellule sensoriali sono immerse nell’endolinfa, il resto è immerso invece nella perilinfa [5]. Quando arriva la stimolazione sonora le cellule cigliate vengono eccitate con il passaggio degli ioni potassio provenienti dall’endolinfa. Questo porta ad una variazione del potenziale di riposo che si trasforma in stimolo elettrico, propagandosi poi alle terminazioni nervose del nervo acustico e quindi ai nuclei del sistema nervoso centrale. Risulta valido anche il processo inverso; infatti terminata la stimolazione sonora, gli ioni potassio vengono poi recuperati dalla perilinfa all’endolinfa [5]. Questo è lo scopo fondamentale che le giunzioni intercellulari rivestono nell’organo dell’udito e che spiega come un loro mutamento porti ad una alterata risposta sensoriale alla stimolazione acustica.
Caratteristiche cliniche
Scott et al. (1995) studiarono una famiglia altamente inbred beduina con autosomica recessiva sordità. La famiglia apparteneva a una tribù fondata circa 200 anni fa da un beduino arabo maschio emigrato dall’Egitto alla regione meridionale di quella che allora era la Palestina. Ha sposato una donna del posto e aveva 7 bambini, 5 dei quali sono sopravvissuti fino all’età adulta. Matrimonio consanguinei era stata la regola della tribù dalla sua terza generazione. La tribù è stata poi nella sua settima generazione e consisteva di circa 3.000 persone, ognuno dei quali residenti in una singola area geografica in Israele che è separata dalle altre comunità beduine. I tassi di natalità all’interno della tribù erano alte, e la poligamia era comune.All’interno della generazione passata c’era stato 80 persone con sordità congenita; tutti gli individui affetti erano discendenti di 2 dei 5 figli adulti del fondatore. La sordità è stata profonda perdita dell’udito neurosensoriale prelinguale con drasticamente elevate soglie audiometriche a tutte le frequenze. Tutti i soggetti avevano un fenotipo sordi altrimenti normale con assenza di anomalie esterne dell’orecchio, retinopatia, o difetti renali, e tutti erano di intelligenza normale. Cheng et al. (2005) ha osservato che il 4% dei 777 bambini non imparentati con perdita di udito aveva cartelle cliniche che hanno elencato una causa ambientale per la sordità, e che l’11% di quelli con eziologia sconosciuta una sono stati trovati ad avere GJB2 / mutazioni GJB6. Test otoemissioni acustiche per rilevare le cellule ciliate esterne funzionali identificati 76 bambini (10%) con emissioni positivi, in linea con neuropatia uditiva. Cinque dei pazienti con neuropatia uditiva erano omozigoti o eterozigoti composti per mutazioni nel gene GJB2. Cheng et al. (2005) suggerivano che la mancanza di giunzioni funzionali dovute a mutazioni GJB2 non necessariamente distruggere tutte le funzionalità delle cellule ciliate esterne. In un sondaggio da Dodson et al. (2011), 127 (54%) dei 235 intervistati con DFNB1dovuta a mutazioni nel GJB2 e / o geni GJB6 riferito disfunzione vestibolare, rispetto al 25 (41%) dei 61 controlli sordi senza DFNB1 sordità (p inferiore a 0,03). La maggior parte dei DFNB1pazienti con vertigine dovuto sdraiarsi per esso a placarsi, e il 48% ha riferito che le vertigini interferito con le attività della vita quotidiana. Vertigo è stato segnalato da significativamente più casi con troncante di nontruncating mutazioni ed è stato anche associato ad una storia familiare di vertigini. Dodson et al. (2011) conclusero che la disfunzione vestibolare è più comune in DFNB1 sordità di quanto precedentemente riconosciuto. Schimmenti et al. (2008)iscritti 95 bambini con perdita uditiva da cui entrambi esoni di Cx26 sono stati sequenziati e la cancellazione Cx30 è stata determinata in uno studio di confronto bambini con e senza perdita di udito connessina-related. Tra i 82 bambini che hanno subito lo screening neonatale, 12 bambini erano passati; 3 avevano perdita dell’udito connessina-related. Non ci sono state differenze nel tasso di screening uditivo neonatale passaggio, complicazione neonatale, o perdita dell’udito gravità tra i bambini con e senza perdita di udito connessina-correlati. Schimmenti et al. (2008) ha sottolineato che non tutti i bambini con sordità connexin legata mancherà lo screening uditivo neonatale. Storia familiare correlata in modo significativo con la perdita di udito connessina-related.
Eredità
Prove genetiche dirette per l’esistenza di almeno 2 non alleliche, recessive, forme fenotipicamente distinguibili di sordità congenita è stato fornito dal pedigree piuttosto frequenti del tipo riportato da Stevenson e Cheeseman (1956). In solo 5 dei 32 sordi ereditaria da accoppiamenti sordi ereditarie erano tutti i bambini sordi. Da questo, gli autori hanno concluso che probabilmente ci sono 6 loci separato per recessive sordità congenita, partendo dal presupposto che i geni mutanti in ogni hanno una frequenza simile. Vedere i commenti di Slatis (1957).Chung et al. (1959) anche sostenuto l’idea di molteplici forme recessive sordità congenita. Fraser (1964) stima che la metà di una grave sordità infantile è dovuto a semplice eredità mendeliana e che l’87% di questo gruppo è autosomica recessiva. In analisi matematica geniale, Morton ( 1960)hanno concluso che ereditarietà recessiva è responsabile per il 68% della sordità congenita, che omozigosi a uno qualsiasi dei 35 loci può risultare in questo fenotipo e che il 16% della popolazione normale sono portatori di un gene per la sordità congenita. Vedere anche Morton(1991). Muhlmann (1930) ha segnalato un caso in cui 2 individui con sordità congenita, chiaramente con malattia autosomica recessiva, perché in ogni caso i genitori erano consanguinee e sib ha risentito, sposato e ha prodotto solo i bambini con udito normale. Mengel et al. (1969) ha presentato un pedigree istruttiva in cui 2 genitori congenitamente sordi avevano tutto normale udito prole. Un genitore è venuto da un gruppo di mennoniti con numerosi casi di sordità congenita in un modello recessivo. L’altro genitore è venuto da un gruppo Amish che conteneva anche più persone con apparentemente ereditaria recessiva sordità congenita.Majumder et al. (1989) ha studiato la genetica della sordità prelinguale in 133 nuclei familiari provenienti da 25 grandi pedigree in India. Segregazione analisi ha rivelato un modello per prelinguale suggestiva sordità scollegato diallelic autosomica loci. Gli individui sono stati colpiti se e solo se erano omozigote recessiva sia loci. In Israele, Brownstein et al. (1991) studiarono famiglie in cui entrambi i genitori avevano sordità congenita. Tra questi 111 coppie in cui la sordità era probabilmente recessiva e c’era almeno 1 figlio, ci sono stati 12 con solo bambini sordi e 5 con entrambi i bambini sordi e udenti. È stato stimato il numero di loci per la sordità recessiva in tutto il gruppo per essere 8 o 9. Accoppiamenti all’interno dello stesso gruppo ebraico (sefardita, orientale, o Ashkenazi) ha dato una stima di 6,7 loci, considerando accoppiamenti interetnici hanno dato una stima di 22 loci. Una conclusione dello studio per la consulenza genetica è che i coniugi sordi provenienti da diversi gruppi etnici hanno un rischio minore per i bambini sordi rispetto a quelli dello stesso gruppo etnico.
Mappatura
Guilford et al. (1994) eseguita linkage analisi usando marcatori microsatelliti altamente polimorfici in 2 famiglie consanguinee dalla Tunisia con sordità profonda prelinguale. A 2 punti lod punteggio massimo di 9.88 a teta = 0.01 è stato trovato con un pennarello sul cromosoma 13q (D13S175). Linkage è stato osservato anche con il pericentromerica D13S115 13q12 loci e D13S143. (Guilford et al. (1994) di cui a questo disturbo come la sordità non sindromica recessiva e utilizzato il simbolo gene NSRD1.) Chaib et al. (1994) studiarono una famiglia di origine francese con una forma autosomica dominante sordità neurosensoriale di. La sordità era da moderata a grave, ha avuto un esordio prelinguale, e colpito prevalentemente le alte frequenze. Con analisi di linkage, hanno mappato il disturbo al cromosoma 13q (multipoint punteggio massimo lod di 4.66 a D13S175). I risultati suggeriva che differenti mutazioni nel gene candidato potrebbe causare sordità neurosensoriale dominanti o recessivi. Questa situazione, con le forme dominanti e recessivi dello stesso disturbo a seconda della natura delle mutazioni specifiche, è stata osservata in epidermolisi bollosa distrofica dovuta a mutazioni nel gene COL7A1 (120120), a retinite pigmentosa dovuta a mutazioni nel gene della rodopsina ( RHO, 180380), in miotonia congenita dovuta a mutazioni nel gene CLCN1 (118425), per elencare solo 3 esempi. Da studi di linkage in 18 Nuova Zelanda e 1 australiano parentele non consanguinei con non sindromica presunta sordità neurosensoriale congenita e una struttura di razza coerente con trasmissione autosomica recessiva, Maw et al. (1995) linkage trovato ai marcatori D13S175, D13S143, e D13S115 sul cromosoma 13. La scoperta ha suggerito che ilDFNB1 locus può dare un contributo importante per la sordità neurosensoriale autosomica recessiva nella popolazione caucasica. Mentre non vi era alcuna prova statisticamente significativa per l’eterogeneità in uno dei loci 3 marcatore testato, 9 delle 19 famiglie hanno mostrato cosegregazione di aplotipi marcatori con sordità. In queste 9 famiglie, variazione fenotipica è stata osservata sia all’interno fratrie (in 4 famiglie), che ha indicato che espressività variabile caratterizzato alcuni genotipi al DFNB1 locus, e tra le generazioni (in 2 famiglie), che ha suggerito l’eterogeneità allelica. Scott et al. (1995) hanno dimostrato che la sordità non sindromica autosomica recessiva in una famiglia beduina altamente inbred era legata al cromosoma 13q12. In 1 di 27 famiglie di origine pakistana con non sindromica sordità recessiva, Brown et al. (1996) trovarono linkage al DFNB1 locus nel cromosoma 13. Le analisi dell’aplotipo di marcatori nella regione di 13q pericentromeric suggerito un evento di ricombinazione che mappato DFNB1 prossimale al D13S175 marcatore e in prossimità di D13S143. In un erratum, gli autori hanno notato che ulteriori analisi collocato D13S143 distale D13S175 anziché prossimale, e quindi il locus DFNB1 é arrivata a trovarsi prossimale a D13S143, come suggerito da Scott et al.(1995). Gasparini et al. (1997) eseguito uno studio di linkage genetico con 4 marcatori microsatelliti legati a DFNB1 in un totale di 48 famiglie mediterranee indipendenti, di cui 30 e 18 erano di origine italiana e spagnola, rispettivamente. Hanno concluso che DFNB1 ha giocato un ruolo nel 79% delle famiglie mediterranee con non sindromica autosomica recessiva sordità neurosensoriale.
Genetica molecolare
Kelsell et al. (1997) identificarono una mutazione omozigote nel gene GJB2 (121011,0002) in membri affetti di 3 famiglie con autosomica recessiva sordità neurosensoriale non sindromica legata alla 13q11-q12 (Brown et al., 1996). Con immunoistochimica, Kelsell et al. (1997)dimostrano che CX26 ha un alto livello di espressione in cellule umane cocleari. Denoyelle et al.(1999) ha studiato 140 bambini da 104 famiglie con vari gradi di perdita dell’udito neurosensoriale. CX26 mutazioni erano presenti in 43 (49%) di 88 famiglie con sordità prelinguale rispetto a nessuno dei 16 famiglie con forme postlinguale di sordità. Sordità variava da lieve a profonda CX26-associato, ed è stato associato con pendenza o curve audiometriche piatti e un normale orecchio interno radiologicamente. La perdita dell’udito non era progressiva in 11 su 16 casi esaminati, e le variazioni della gravità della sordità tra fratelli e sorelle erano comuni. Denoyelle et al. (1999) suggerivano che un elemento importante per la consulenza genetica è che la gravità della perdita di udito in DFNB1 è estremamente variabile e non può essere previsto, anche all’interno delle famiglie. Dahl et al. (2006) identificarono una mutazione omozigote nel gene GJB2 (V37I; 121011,0023) a 4 (8,3%) dei 48 bambini australiani con lieve o lieve perdita dell’udito neurosensoriale. Tutti e 4 i bambini erano a fondo asiatico, e l’analisi SNP hanno suggerito un effetto comune fondatore. Tutti i 4 bambini hanno mostrato sordità neurosensoriale bilaterale ad alta frequenza, e 3 avevano anche perdita di udito a bassa frequenza. Due altri bambini che erano eterozigoti per V37I avevano lieve ad alta frequenza di perdita massima a 6 kHz, e la perdita di bassa frequenza mite, rispettivamente. In tutto, 55 bambini con lieve o lieve perdita di udito sono stati identificati in una proiezione di 6.240 bambini delle scuole australiane. Tang et al. (2006) analizzato il gene GJB2 in 610 soggetti con problemi di udito e 294 controlli e mutazioni patogenetiche identificate nel 10,3% dei casi, con risultati equivoci a 1,8% dei casi a causa della rilevazione di non classificati, romanzo, o controverse variazioni di sequenza di codifica o di solo una singola mutazione recessiva in GJB2. Tredici variazioni di sequenza sono stati identificati nei controlli, e genotipi complessi sono stati osservati tra i controlli asiatici, 47% dei quali portato da 2 a 4 variazioni di sequenza nella regione codificante del gene GJB2. Iossa et al. (2010) ha riferito una famiglia italiana in cui un non colpiti madre e 1 dei suoi figli sordi erano entrambi eterozigoti per un allele portando 2 mutazioni GJB2 in cis: il R75Q dominante (121011,0026) e il recessivo 35delG (121011,0005),mentre l’altro suo figlio sordo non ha effettuato una di queste mutazioni. I risultati suggeriscono che la mutazione recessiva ‘annullato’ l’effetto della mutazione dominante provocando una proteina troncata prima di raggiungere residuo 75. Iossa et al. (2010) suggerivano che la sordità nei 2 figli era dovuta ad altre cause genetiche e sottolineato l’importanza della relazione per la consulenza genetica. La sordità, DIGENICA, GJB2 / GJB6 Del Castillo et al. (2002) notarono che in molti pazienti (10-42%) con autosomica recessiva sordità sindromica che sono stati trovati ad avere una mutazione nel gene GJB2, la seconda mutazione è rimasto non identificato. Essi hanno dimostrato che 22 dei 33 non imparentati questi pazienti, 9 dei quali avevano prova di linkage a 13q12, erano eterozigoti composti per una mutazione nel gene GJB2 (35delG; 121011,0005) e una delezione nel gene GJB6 (604418,0004). Due soggetti erano omozigote per la mutazione GJB6.Nella popolazione spagnola, l’eliminazione GJB6 è stato il secondo più frequente mutazione che causa la sordità prelinguale. Gli autori conclusero che mutazioni nel gene GJB2 e GJB6 può risultare in un modello monogenica o DIGENICA di eredità della sordità prelinguale. Del Castillo et al. (2002) riportarono la cancellazione come 342 kb, ma Del Castillo et al. (2005) ha affermato che i dati di sequenziamento più recenti indicano che l’eliminazione è di 309 Kb.Pallares-Ruiz et al. (2002) trovarono una delezione nel gene GJB6 in trans in 4 dei pazienti 6 sordità eterozigoti per una mutazione GJB2, suggerendo un modo DIGENICA di eredità. In 4 pazienti spagnoli non imparentati con autosomica insufficienza uditiva non sindromica recessiva che erano eterozigoti per 1 GJB2 allele mutante e non ha effettuato la GJB6 309-kb delezione, del Castillo et al. (2005) identificarono una delezione GJB6 232-kb, che hanno indicato come del (GJB6-D13S1854) (vedi 604418,0006). La soppressione è stata successivamente trovata in DFNB1 pazienti nel Regno Unito, Brasile, e l’Italia settentrionale; analisi dell’aplotipo ha rivelato un fondatore comune condiviso tra i cromosomi studiati dalla Spagna, il Regno Unito, e in Italia. Nei 255 pazienti francesi con un fenotipo compatibile con DFNB1, Feldmann et al.(2004) ha rilevato che il 32% ha avuto mutazioni GJB2 biallelic, e il 6% era eterozigoti composti per una mutazione GJB2 e il GJB6 342-kb delezione. Profondamente bambini sordi avevano più probabilità di avere il GJB2 biallelic o eterozigoti GJB2 / mutazioni GJB6. In uno studio di 777 bambini non imparentati con perdita di udito, Cheng et al. (2005) GJB2 o mutazioni GJB6 nel 12% identificato; tra quelli con un colpiti sib, il 20% ha avuto mutazioni GJB2 o GJB6. Dieci pazienti erano eterozigoti composti per mutazioni nel GJB2 e geni GJB6. In 324 probandi con perdita di udito e 280 controlli, tra cui 135 probandi e 280 controlli precedentemente riportati daTang ed altri. (2006), Tang et al. (2008) screening per variazioni di sequenza del DNA in GJB2 e delezioni in GJB6. Il 232-kb delezione GJB6 non è stato trovato, e il 309-kb delezione GJB6 è stato trovato solo una volta, in un paziente di sconosciuta origine etnica che era anche eterozigoti per una mutazione nel GJB2 tronca. Tang et al. (2008) suggeriva che i 232- e 309-kb delezioni nel gene GJB6 non possono essere comuni in tutte le popolazioni. Sordo, DIGENICA, GJB2 / GJB3Liu et al. (2009) riportarono DIGENICA ereditarietà della sordità non sindromica causata da mutazioni nel GJB2 e GJB3 (603324) geni. Tre di 108 probandi cinesi con autosomica recessiva sordità e solo 1 allele mutante GJB2 (ad esempio, 121011,0014) sono risultati essere composti eterozigoti con una mutazione GJB3 (603324,0011; 603324,0012). I risultati erano coerenti con ereditarietà DIGENICA; i genitori non affetti erano eterozigoti per 1 degli alleli mutanti.Recensioni Willems (2000) ha esaminato le cause genetiche della sordità neurosensoriale non sindromica. Petersen e Willems (2006) hanno fornito una dettagliata revisione della genetica molecolare della sordità non sindromica autosomica recessiva. ![]()
Genetica di popolazione
In Tunisia, Ben arabo et al. (1990) stimato la frequenza di autosomica recessiva non sindromica sordità neurosensoriale di essere 7 per 10.000. Chaabani et al. (1995) hanno studiato 30 coppie sordi in Tunisia e ha stimato che il numero di loci per autosomica recessiva non sindromica sordità in questa popolazione era 8.3. Nance et al. (2000) ha proposto una ipotesi per l’alta frequenza di DFNB1 in molte grandi popolazioni del mondo, sulla base di un’analisi della proporzione di matrimoni noncomplementary tra sordi corso del 19 ° secolo, che ha suggerito che la frequenza di DFNB1 essere raddoppiata negli Stati Uniti nel corso degli ultimi 200 anni.Questi cosiddetti matrimoni noncomplementary tra individui con lo stesso tipo di sordità recessiva sono incapaci di produrre dell’udito prole, e la radice quadrata della loro frequenza tra matrimoni sordi fornisce un limite superiore per la prevalenza della forma più comune di sordità recessiva in quel momento . Per spiegare l’aumento, hanno suggerito che la combinazione di intensa accoppiamento assortative e selezione rilassata aumentato sia il gene e le frequenze dei fenotipi per DFNB1. Il modello proposto dal presupposto che in millenni precedenti l’idoneità genetica degli individui con sordità profonda congenita era molto basso e che i geni per la sordità sono stati poi in un equilibrio mutazione. L’introduzione della lingua dei segni in Europa nel 17 al 18 ° secolo è stato un evento chiave che ha migliorato notevolmente le condizioni sociali ed economiche dei sordi, insieme con la loro idoneità genetica. In molti paesi, sono state stabilite le scuole per sordi, contribuendo alla comparsa di omogamia linguistico intenso, vale a dire, la selezione del partner in base alla capacità di comunicare in lingua dei segni. In alcune grandi popolazioni, connessina-26 sordità è stato osservato, ma in modo molto bassa frequenza. In Mongolia, per esempio, dove c’è solo 1 scuola residenziale per i sordi, linguaggio dei segni è stato introdotto solo 1995. Inoltre, l’idoneità dei sordi è molto inferiore a quello dei loro fratelli e sorelle di udito, l’accoppiamento assortative è molto meno frequente che in gli Stati Uniti, e le mutazioni connessina rappresentano solo il 1,3% di tutta la sordità (Pandya et al., 2001). Nance e Kearsey (2004) ha dimostrato mediante simulazione al computer che assortative mating, infatti, in grado di accelerare notevolmente la risposta genetica alla selezione rilassato . Insieme con gli effetti della deriva genetica e consanguineità, accoppiamento assortative anche può aver giocato un ruolo chiave nell’evoluzione comune e accelerare la fissazione di geni per discorso dopo la loro prima apparizione in Homo sapiens 100.000 a 150.000 anni fa. Nei 156 pazienti Repubblica congenitamente sordi indipendenti ,Seeman et al. (2004) testato per la presenza di mutazioni nella sequenza codificante del gene GJB2. Almeno 1 mutazione patogenicità in 48,1% dei pazienti. Le 3 mutazioni più comuni erano W24X (121011,0003), 35delG (121011,0005), e 313del14 (121011,0034); gli autori hanno affermato che i test per solo questi 3 mutazioni sarebbe rilevare oltre il 96% di tutte le mutazioni che causano la malattia in GJB2 in questa popolazione. Test per 35delG in 503 controlli hanno rivelato una frequenza portante di 1:. 29,6 (3,4%), nella Repubblica Ceca Alvarez et al. (2005) uno screening del gene GJB2 in 34 spagnoli Romani (zingara) famiglie con autosomica recessiva perdita di udito non sindromica e trovarono mutazioni nel 50%. L’allele predominante era W24X (121011,0003), pari al 79% di DFNB1 alleli. Le analisi di aplotipo suggerito che un effetto fondatore è responsabile per l’alta prevalenza di questa mutazione tra gitani spagnoli. 35delG(121011,0005) è stato il secondo allele più comune (17%). Arnos et al. (2008) ha raccolto dati su 311 matrimoni pedigree contemporanei tra gli individui sordi che erano paragonabili a quelli raccolti da Fay (1898). Segregazione analisi dei dati ottenuti è emerso che la percentuale stimata di accoppiamenti noncomplementary che possono produrre solo i bambini sordi aumentato di un fattore superiore a 5 in quanto detto 100 anni. Ulteriori analisi all’interno del loro campione di pedigree contemporanei ha dimostrato che vi è stato un aumento statisticamente significativo lineare della prevalenza delle mutazioni GJB2 patologiche quando i dati su 441 probandi sono stati suddivisi in tre coorti di nascita di 20 anni (1920-1980). Arnos et al. (2008) hanno concluso che i loro dati erano coerenti con l’aumento della frequenza di DFNB1 previsto dai loro studi di simulazione precedenti, e dimostrato convincente per l’influenza importante che mating assortative può avere sulla frequenza dei geni comuni per la sordità. Schimmenti et al. (2008)iscritti 95 bambini con perdita uditiva da cui entrambi esoni di Cx26 sono stati sequenziati e la cancellazione Cx30 è stata determinata in uno studio di confronto bambini con e senza perdita di udito connessina-related. Complessivamente tra questi 95 pazienti, biallelic mutazioni sono state identificate nel 24,7%, ma solo nel 9,1% dei bambini di origine ispanica. Schimmenti et al.(2008) conclusero che la perdita di udito connessina-simile si verifica in un quarto dei bambini in una popolazione etnicamente diversificata perdita di udito, ma con una minore prevalenza nei bambini ispanici. Tekin et al. (2010) uno screening del gene GJB2 in 534 probandi mongole con non sindromica sordità neurosensoriale e identificate mutazioni GJB2 biallelic in 23 (4,5%) probandi sordi. La mutazione più comune, IVS1 + 1G-A (121011,0029), sembrava avere origini diverse in base a più aplotipi associati. Tekin et al. (2010) ha dichiarato di aver trovato una minore frequenza di accoppiamento assortative (37,5%) e una diminuzione di fitness genetica (62%) dei sordi in Mongolia, rispetto alle popolazioni occidentali, che spiegava la frequenza più bassa della sordità GJB2 in Mongolia. Barashkov et al. (2011) trovarono omozigosi per la IVS1 + 1G-A mutazione in GJB2 in 70 su 86 pazienti dalla popolazione Yakut nella Siberia orientale isolare con insufficienza uditiva non sindromica. Sei pazienti erano eterozigoti composti per questa mutazione e un’altra mutazione patogena GJB2. Esame audiometrico è stato eseguito su 40 pazienti che erano omozigote per la mutazione. La maggior parte (85%) hanno avuto una grave insufficienza di uditiva profonda, il 14% ha avuto danno moderato, e l’1% ha avuto una perdita dell’udito lieve. C’era una certa variabilità nelle soglie uditive. La frequenza portante per questa mutazione in questa popolazione è stata stimata essere dell’11,7%, il più alto tra 6 popolazioni siberiane orientali analizzati, e la mutazione è stato stimato in circa 800 anni. I risultati erano coerenti con un effetto fondatore, e Barashkov et al. (2011) postulato una origine asiatica centrale per la mutazione. Tra 15.799 etnicamente diversi individui a screening perDFNB1 stato di portatore, Lazarin et al. (2013) identificarono 371 vettori (2,3%), per una frequenza stimata portante di circa 1 a 43. Cinque coppie ‘carrier’ sono stati identificati. Sei individui sono stati identificati come omozigoti o eterozigoti composti. Tra 756 individui di origine asiatica orientale, la frequenza portante era di 1 a 22. ![]()
Storia
Nell’era pre-mendeliana, Meniere (1846, 1856) ha rilevato il ruolo della consanguineità dei genitori sordità. Boudin (1862) ha rilevato l’associazione tra la consanguineità e la sordità congenita. Groce (1985) tracciato la storia della sordità congenita a Martha Vineyard, il isola del Massachusetts. La prima persona sorda spostato l’isola nel 1694. Groce (1985) ha stimato che nel 19 ° secolo 1 a 155 persone sull’isola è nato sordo. Perché c’erano membri sordi in quasi ogni famiglia nella parte occidentale dell’isola, ognuno ha imparato il linguaggio dei segni, ei sordi sono stati pienamente integrata in ogni aspetto della vita. In queste circostanze, la sordità non era una disabilità o un handicap. Mengel et al. (1967) trovarono una grave sordità in 16 membri di un gruppo di affini. Dalla storia, tutti sono nati con almeno un po ‘udienza ma ha subito una grave perdita progressiva nella tarda infanzia. Analisi ecografica e discorso ha dato un’ulteriore prova di un po ‘di udito nella prima infanzia. Test audiologica suggerito posizione cocleare del difetto. Anche se le generazioni successive sono state colpite in alcuni casi, la consanguineità ed ereditarietà recessiva sono stati pensati per spiegare il ritrovamento. Barr e Wedenberg (1964)descrivono un disturbo simile a 4 su 7 fra fratelli e sorelle. Tra i 11 figli di genitori consanguinei,Cremers (1979) ha osservato 2 ragazzi e una ragazza con progressiva sordità neurosensoriale, prima notato alle età di 4, 7 e 11 anni. Ha trovato 2 segnalazioni di una sordità simile e ha concluso che era diversa da quella sordità riportata da Mengel et al. (1967). Una seconda famiglia è stato riportato da Cremers et al. (1987). Progressiva ipoacusia neurosensoriale iniziata soprattutto nelle frequenze più alte. Hanno anche trovato un brusco calo del audiogramma che lentamente diminuita con l’aumento di perdita dell’udito a bassa frequenza. Ormerod (1960) ha riconosciuto i seguenti tipi di sordità congenita, a cominciare con la forma più completa: (1) tipo Michel – completa mancanza di sviluppo di orecchio interno. (2) Tipo di Mondini-Alexander – lo sviluppo di una sola tubo ricurvo che rappresenta la coclea, e simili l’immaturità dei canali vestibolari. (3) Tipo di Bing-Siebenmann – labirinto osseo ben formate ma parte membranosa e in particolare l’organo di senso poco sviluppato. Questo tipo è spesso associato con retinite pigmentosa. (4) Scheibe tipo cochleosaccular – In questa forma, che è quella più frequente, la parte vestibolare è sviluppato e funzionante. Malformazione è limitato alla coclea membranosa e saccule. Questo tipo si verifica nella sindrome di Waardenburg. (5) tipo Siebenmann – cambia soprattutto nel dell’orecchio medio e spesso a causa di deficit di ormone tiroideo. L’orecchio medio è coinvolta nel cambiamento mixomatosa che può essere persistenza embrionale. (6) Microtia e atresia del meato – anomalia limitato all’orecchio esterno.
II° approfondimento
· GeneReviews Ricerca Avanzata
· Aiuto
Sordità Non sindromica, , DFNB1
Sinonimo: GJB2 -related DFNB 1 non sindromica, udito e sordità
Richard JH Smith, MD e Guy Van Camp, PhD.
Distacco iniziale: 28 settembre 1998; Ultimo aggiornamento: 2 gennaio 2014.
Sommario
Caratteristiche cliniche.
Perdita di udito non sindromica e sordità (DFNB1) è caratterizzata da congenita, non progressivo, insufficienza uditiva neurosensoriale da lieve a profonda. Non sono emerse altre mediche associate sono presenti.
Diagnosi / testing.
La diagnosi di DFNB1 dipende test di genetica molecolare per identificare le varianti che causano sordità-in GJB2 e monte cis elementi -regulatiory che alterano la beta-2 proteina gap junction (connessina 26). Test di genetica molecolare di GJB2 rileva oltre il 99% delle varianti-sordità causando in questi geni.
Gestione.
Trattamento delle manifestazioni: Apparecchi acustici; l’iscrizione in appositi programmi di istruzione;l’impianto cocleare può essere considerato per gli individui con sordità profonda.
Sorveglianza: Sorveglianza include esami annuali e ripetere audiometria per dimostrare la stabilità di perdita dell’udito.
La valutazione dei parenti a rischio: se entrambe le varianti-sordità causa sono stati identificati in un colpiti membro della famiglia, test di genetica molecolare in grado di chiarire lo status genetico di un bambino che può avere DFNB1 in modo che possono essere forniti adeguato supporto e la gestione precoce.
Consulenza genetica.
DFNB1 è ereditata come autosomica recessiva maniera. In ogni gravidanza, i genitori di unprobando hanno una probabilità del 25% di avere un bambino sordo, una probabilità del 50% di avere un figlio un’udienza che è un elemento portante, e una probabilità del 25% di avere un figlio un’udienza che non è un vettore.Una volta che un sib a rischio è noto per essere udito, la possibilità della sua / suo essere un vettore è 2/3. Quando le varianti che causano DFNB1 vengono rilevate in un membro della famiglia, il test del vettore per i familiari a rischio e test prenatale per gravidanze a rischio sono possibili.
Diagnosi
Diagnosi clinica
Perdita dell’udito e sordità non sindromica (DFNB1) è associato con il seguente:
- Congenita, generalmente non progressiva insufficienza uditiva neurosensoriale che è mite di profonda da cervello uditivo test risposta stelo (ABR) o puro tono audiometria Nota: (1) Audizione si misura in decibel (dB).La soglia o 0 dB contrassegno per ogni frequenza si riferisce al livello al quale normali giovani adulti percepiscono un Tone Burst 50% del tempo. Audizione è considerato normale se le soglie di un individuo sono a 25 dB di soglie normali. (2) La gravità della perdita dell’udito è classificato come lieve (26-40 dB), moderata (41-55 dB), moderatamente grave (56-70 dB), grave (71-90 dB), o profonda (90 dB). La frequenza di perdita dell’udito è designato come basso (<500 Hz), medio (501-2000 Hz), o alto (> 2000 Hz) (vedi sordità ereditaria e dell’udito Panoramica perdita).
- Nessun risultati sistemici correlati identificati da anamnesi e l’esame obiettivo
- Una storia familiare di perdita di udito non sindromica coerente con autosomica recessiva ereditarietà
Molecolare Test genetici
. Gene GJB2, che codifica connessina 26, è l’unico gene in cui mutazione è noto per provocare la sordità al DFNB1locus:
- GJB2. Circa il 98% degli individui con DFNB1 hanno due identificabili GJB2 varianti (cioè, sono omozigoti o eterozigoti composti). Più della metà di tutte le persone di discendenza nord-europea con due identificabili GJB2varianti sono omozigote per il singolo c.35delG nucleotide variante [Scott et al 1998]. I dati per quanto riguarda l’associazione della GJB2 varianti p.Met34Thr e p.Val37Ile con DFNB1 sono discussi in Genetica Molecolare.
Circa il 2% degli individui con DFNB1 hanno uno identificabile GJB2 variante e una delle tre grandi delezioni comprese le sequenze a monte di GJB2 e una porzione di GJB6. L’eliminazione provocano ridotta espressa della valleGJB2 gene, presumibilmente a causa della cancellazione di un cis elemento -regulatory. Inizialmente questo coinvolgimento di parziale GJB6 delezione era considerato un esempio di eredità DIGENICA che l’inattivazione diGJB2 su un allele e l’inattivazione di GJB6 sul secondo. Recenti evidenze confermano che DIGENICA ereditarietà coinvolgono questi due geni non è un fattore nella patogenesi (vedi Genetica Molecolare)
Sperimentazione clinica
- Le analisi della sequenza. Analisi di sequenza di dell’esone 2, che ospita l’intera regione codificante, rileva entrambe le varianti nel 98% delle persone con DFNB1. Analisi Variant possono includere il sequenziamento di ulteriori varianti rare, tra cui l’esone 1 variante di sito di splice e le eliminazioni a monte che coinvolgono GJB6(vedi Genetica Molecolare).
Tabella 1.
Sintesi di ricerca di genetica molecolare Utilizzato in DFNB1
|
Gene 1 |
Metodo di prova |
Allelic Varianti rilevato 2 |
Rilevazione Variant frequenza con il metodo di prova di 3 |
|
|
Due varianti |
Una variante |
|||
|
GJB2 |
Le analisi della sequenza 4 |
GJB2 varianti di sequenza 5 |
98% |
~ 2% 6 |
|
Cancellazione /duplicazione analisi 7 |
<< 1% |
<< 1% |
||
|
Analisi mirata per varianti patogeniche |
Soppressione di GJB2 elementi regolatori a monte 9 |
N / A |
~ 2% 6 |
|
NA = non applicabile
1.Vedi Tabella Geni A. e database per cromosoma locus e proteine.
2.Vedere Genetica Molecolare per informazioni su GJB2 varianti.
3.La capacità del metodo utilizzato per rilevare una variante che è presente nella indicata gene
4.Le analisi della sequenza rileva le varianti che sono benigni, probabilmente benigno, di significato incerto, probabilmente patogeni o patogeni. Varianti patogeniche possono includere piccole intragenica delezioni / inserzioni e missense, nonsense, e varianti sito di splicing; tipicamente, esone o all’in- grosso gene delezioni / duplicazioni non vengono rilevati. Per problemi da considerare nell’interpretazione di analisi di sequenza risultati, fare clic qui.
5.Alcune varianti hanno un pregiudizio etnico: c.35delG è più comune nelle popolazioni di origine nord-europea; c.167delT è più comune nel ashkenazi popolazione, c.235delC è più comune nelle popolazioni giapponesi e cinesi.
6.Percentuali variano a seconda etnia. I numeri in tabella riflettono lo screening di una popolazione degli Stati Uniti in primo luogo di discendenza nord-europea.
7.Test che identifica esone o all’in- grosso gene delezioni / duplicazioni non facilmente rilevabili mediante analisi della sequenza di codifica e regioni fiancheggianti intronic di genomico del DNA; inclusi nella varietà di metodi che possono essere utilizzati sono:PCRquantitativa, a lungo raggio PCR, multiplex ligation-dipendente probe amplificazione (MLPA), e cromosomica microarray(CMA) che include questo / gene cromosomico segmento.
8.Vedere Test Strategia e tabella 3.
9.Feldmann et al [2009] hanno riportato una contigua gene delezione che comprendeva GJB2 e due connexine contigui, GJA3 e GJB6, oltre ad una porzione di CRYL1 in trans con un noto GJB2 variante sordità provocando in un individuo con profonda perdita dell’udito prelinguale, ritardo mentale e psicomotorio sviluppo, clinodattilia del secondo dita dei piedi, e un ciuffo frontale[Feldmann et al 2009].
Interpretazione dei risultati delle prove
- La diagnosi di DFNB1 si stabilisce se un individuo o colpiti fratello ha riconosciuto varianti-sordità causando inGJB2 o in note regioni regolatorie del gene.
- Se solo una GJB2 viene rilevato variante e una grande delezione di nota regione regolatrice upstream (che include una porzione di GJB6) non è presente, l’interessato individuo è o: (1) sordi e casualmente un vettore di unGJB2 variante o (2) sordi con DFNB1 secondaria ad un non-romanzo GJB2, variante non complementare nel DFNB1 dell’intervallo. Nota: E ‘difficile determinare la percentuale delle persone sorde con un GJB2 variante che rientrano in queste due categorie. In una schermata di individui sordi eterozigoti per c.35delG, analisi dei single nucleotide polimorfismi (SNP) nel GJB2-GJB6 regione sostiene con forza l’esistenza di nuove varianti nell’intervallo DFNB1 in alcuni di questi individui [Azaiez et al 2004, del Castillo et al 2005].
Strategia dei saggi
Per confermare / a stabilire la diagnosi in un probando. Per gli individui sospettati di avere DFNB1:
Una strategia di sperimentazione è l’analisi di GJB2:
- Il primo passo è l’analisi di sequenza di GJB2 esone 2 (la regione codificante di GJB2). Se vengono individuati due varianti-sordità causa, viene stabilita la diagnosi di DFNB1.
- Se viene identificata una variante-sordità che causano, analisi mirate per uno dei due grandi delezioni note di monte di GJB2 (e compresi GJB2 sequenze regolatrici e GJB6), ΔGJB6-D13S1830 e ΔGJB6-D13S1854, è garantito.
- Se non varianti-sordità causa di GJB2 sono identificati, analisi mirate per l’eliminazione di un GJB2 sequenza di regolamentazione a monte non è giustificata. La frequenza di queste delezioni in tutte le popolazioni non è sufficientemente alta da provocare un gran numero di individui sordi omozigoti per queste varianti. Essi rappresentano meno dello 0,5% di tutte le persone con sordità prelinguale e senza varianti in GJB2 [Del Castillo et al 2003, del Castillo et al 2005, Wilch et al 2010].
Una strategia alternativa test genetici è l’uso di un multi-gene pannello che include GJB2 e altri geni di interesse (vedi Diagnosi differenziale). Nota: I geni inseriti e metodi utilizzati in pannelli multi-gene variano da laboratorio e nel tempo.
Geneticamente Correlate (alleliche) Disturbi
DFNA3 è un autosomica dominante disturbo progressivo, da moderata a grave compromissione neurosensoriale; è causata da mutazioni di GJB2 o (meno frequentemente) GJB6.
Cheratoderma palmoplantare con la sordità è un autosomica dominante disturbo causato da mutazioni di GJB2 e caratterizzata da ipercheratosi diffusa delle mani e dei piedi [Richard et al 1998, Heathcote et al 2000].
Cheratite-ittiosi-sordità (KID) La sindrome è una displasia ectodermica in cui influenzato gli individui hanno vascolarizzante cheratite, progressivo erythrokeratoderma e profonda perdita dell’udito neurosensoriale, così come alopecia cicatriziale e predisposizione al carcinoma a cellule squamose [Richard et al 2002, van Geel et al 2002, van Steensel et al 2002]. La sindrome KID è causata da eterozigoti di mutazione di GJB2.
La sindrome Hystrix come ittiosi-sordità (HID) è un autosomica dominante disturbo cheratinizzanti caratterizzata da ipoacusia neurosensoriale e ipercheratosi della pelle. Poco dopo la nascita, eritrodermia sviluppa, con ipercheratosi appuntita e ciottoli, come di tutta la superficie della pelle che appaiono dall’età di un anno. Grave cheratoderma palmo-plantare e alopecia cicatriziale si verificano in alcuni. Sindrome HID è considerato diverso dalla sindrome KID in: (1) la misura e il tempo di comparsa di sintomi cutanei; (2) la gravità della cheratite; e (3) di elettroni caratteristiche microscopiche. KID e HID sindromi sono causati dalla stessa variante patogena in GJB2 [van Geel et al 2002].
La sindrome Vohwinkel è un autosomica dominante condizione classificata come “mutilazione” cheratoderma diffusa a causa ipercheratosi circonferenziale delle cifre può portare a autoamputazione. Lieve a moderata ipoacusia neurosensoriale è spesso associata con la malattia [Maestrini ed altri 1999].
Nota: La variante p.Met34Thr descritta in una famiglia con cheratoderma palmo-plantare e autosomica dominantesordità neurosensoriale [Kelsell et al 1997] non è una causa di perdita dell’udito dominante [Cucci et al 2000]. Questo stesso DNA variante è stata individuata in persone udito normale [Denoyelle et al 1998, Kelley et al 1998], e uno schermo di 128 nonni o capi di singole famiglie non noti per essere connessi e inclusi nel CEPH (Centre d’Etude du Polymorphisme Humain) ha identificato tre persone (2,3%) con la variante [dati non pubblicati]. La possibile patogenicità di p.Met34Thr rimane controverso [Snoeckx et al 2005].
Con alcune varianti nel GJB2, la malattia epidermica e perdita dell’udito cosegregate, mentre altre varianti, la gravità della malattia fenotipo varia, suggerendo che altri fattori modificano gene expression [Kelsell et al 2001].
Caratteristiche cliniche
Descrizione clinica
Perdita di udito non sindromica e sordità (DFNB1) è caratterizzata da congenita (presente alla nascita), non progressiva insufficienza uditiva neurosensoriale. Variabilità intrafamiliare nel grado di sordità è visto.
- Se un affetto persona ha la sordità grave a profonda, un fratello presentare gli stessi GJB2 varianti-sordità provocando ha una probabilità del 91% di avere gravi a profonde sordità e una probabilità del 9% di avere la sordità lieve-moderata.
- Se un affetto persona ha sordità lieve a moderata, un fratello presentare gli stessi GJB2 varianti-sordità provocando ha una probabilità del 66% di avere lieve-moderata la sordità e una probabilità del 34% di avere la sordità grave a profonda.
- Alcuni rapporti descrivono i bambini con GJB2 varianti che superato lo schermo uditivo neonatale e aveva perdita di udito po ‘più tardi insorgenza [Norris et al 2006, Orzan e Murgia 2007].
In una grande analisi trasversale di GJB2 genotipo e dati audiometrici da 1531 individui con autosomica recessiva, da lieve a profonda, sordità non sindromica (età media 8 anni; il 90% entro l’età 0-26 anni) provenienti da 16 paesi, analisi di regressione lineare dell’udito soglie dell’età nell’intero studio e in sottogruppi definiti da genotipo non hanno mostrato significativa progressione della perdita di udito in ogni individuo [Snoeckx et al 2005]. Questo risultato è in accordo con studi precedenti [Denoyelle et al 1999, Orzan et al 1999, Loffler et al 2001]; tuttavia, la progressione di perdita dell’udito non può essere escluso definitivamente data la natura trasversale delle analisi di regressione. Snoeckx et al [2005] hanno trovato un leggero grado di asimmetria, anche se la differenza in media tono puro a 0.5, 1.0, e 2.0 kHz tra orecchie era meno di 15 dB nel 90% degli individui.
Funzione vestibolare è normale; colpite lattanti e bambini non sperimentano problemi di equilibrio e imparare a sedersi e camminare, a volte adatti alla loro età.
Fatta eccezione per il deficit uditivo, colpiti gli individui sono in buona salute; durata della vita è normale.
Genotipo-fenotipo Correlazioni
Numerosi studi hanno dimostrato che è possibile prevedere fenotipo sulla base del genotipo. Il più grande studio finora ha coinvolto un’analisi trasversale di GJB2 genotipo e dati audiometrici da 1531 persone provenienti da 16 paesi diversi, con autosomica recessiva, da lieve a profonda, sordità sindromica [Snoeckx et al 2005]. Tra le 83 diverse varianti identificate, 47 sono stati classificati come non-inattivante (ad esempio, le varianti missenso) e 36 come inattivazione (ad esempio, codoni di stop prematuri). Classificando le varianti in questo modo, gli autori definiscono tre classi genotipiche:
- Inattivante biallelic (/ I I) varianti. 1183 delle 1531 persone studiate (77,3%) segregate due varianti inactivating che rappresentavano 64 diversi genotipi (36% di tutti i genotipi trovato). Il grado di compromissione dell’udito in questa coorte è stata: profonda nel 59% -64% degli individui; grave nel 25% -28%; moderato nel 10% -12%; e mite in 0% -3%.
- Biallelic non inattivante (NI / NI) varianti. Novantacinque delle 1531 persone studiati (6,2%) segregati due varianti non inattivanti che rappresentavano 42 diversi genotipi (24% di tutti i genotipi trovato). Il grado di compromissione dell’udito era mite nel 53% degli individui e grave a profonda nel 20% degli individui.
- Composto inattivante eterozigoti / non-inattivante (I / NI) varianti. Dei 1531 soggetti studiati, 253 (16,5%) segregato uno inattivante (I) e uno (NI) variante non inattivanti che rappresentava 71 differenti genotipi (40% di tutti genotipi trovati). Il grado di ipoacusia è stato profondo nel 24% -30% degli individui e grave nel 10% -17% degli individui.
Diagrammi a dispersione sono stati costruiti per mostrare la media media tono puro binaurale (PTA) a 0,5, 1, e 2 kHz (PTA 0.5,1,2kHz) per ogni persona all’interno di ogni genotipo classe, utilizzando individui omozigoti per la c.35delG allelecome gruppo di riferimento:
- . I / I Solo due genotipi differivano significativamente dal c.35delG omozigoti gruppo di riferimento:
- Individual doppiamente eterozigoti per [GJB2: c.35delG] + [GJB6: del (GJB6 -D13S1830)] era significativamente maggiore deficit uditivo (mediana PTA 0.5,1,2kHz = 108 dB; p <0.0001).
- Gli individui che sono GJB2 eterozigoti composti per [c.35delG] + [- 3179G> A, conosciuta anche come IVS1 + 1G → A] era significativamente meno deficit uditivo (mediana PTA 0.5,1,2kHz = 64 dB; p <0.0001).
- . I / NI Nove genotipi differivano significativamente dal c.35delG omozigoti gruppo di riferimento:
- Un GJB2 composti eterozigoti genotipo, [c.35delG] + [p.Arg143Trp], ha mostrato significativamente maggiore deficit uditivo.
- Otto genotipi erano significativamente meno deficit uditivo. I tre genotipi con il deficit uditivo almeno erano GJB2 eterozigoti composti [c.35delG] + [p.Val37Ile] (PTA mediana 0.5,1,2kHz = 40 dB, p <0,0001), [c.35delG] + [p.Met34Thr ] (PTA mediana 0.5,1,2kHz = 34 dB, p <0,0001), e doppi eterozigoti [Δ GJB6 -D13S1830] + [GJB2: p.Met34Thr] (mediana PT A0.5,1,2kHz = 25 dB, p <0,0001). La scoperta nella classe genotipica T / NT per quanto riguarda la distribuzione di soglia in persone con [c.35delG] + [p.Leu90Pro] ha suggerito una distribuzione bimodale, come sette [c.35delG] + [p.Leu90Pro] GJB2 eterozigoti composti avevano un PTA 0.5,1,2kHz superiore a 95 dB e 34 aveva una PTA 0.5,1,2kHz inferiore a 65 dB, con la PTA 0.5,1,2kHz di un solo individuo cade tra questi due valori (65-95 dB).
- . NI / NI Tre genotipi differiva significativamente dal c.35delG omozigote gruppo di riferimento di avere meno problemi di udito:
- omozigoti p.Met34Thr (PTA mediana 0.5,1,2kHz = 30 dB, p <0,0001)
- omozigoti p.Val37Ile (PTA mediana 0.5,1,2kHz = 27 dB, p <0,0001)
- [p.Met34Thr] + [p.Val37Ile] eterozigoti composti (PTA mediana 0.5,1,2kHz = 23 dB, p <0,001)
Nomenclatura
DFNB seguita da un numero intero suffisso viene utilizzato per designare loci per autosomica recessiva sordità non sindromica.
Prevalenza
DFNB1 rappresenta circa il 50% dei congenita, grave a profonda, autosomica recessiva perdita di udito non sindromica negli Stati Uniti, Francia, Gran Bretagna e Nuova Zelanda / Australia [Denoyelle et al 1997, Green et al 1999]. La sua prevalenza approssimativa nella popolazione generale è 14: 100.000, in base al seguente calcolo: l’incidenza di indebolimento dell’udito congenita ereditaria è 1: 2000 neonati, di cui il 70% hanno perdita di udito non sindromica.Settantacinque al 80% dei casi di perdita di udito non sindromica è autosomica recessiva; di questi, il 50% risultato damutazione di GJB2. Così, 5: 10.000 x 0,7 x 0,8 x 0,5 = 14: 100.000.
Data l’estrema eterogeneità dei autosomica recessiva insufficienza uditiva non sindromica, non è sorprendente che gli studi epidemiologici in altre popolazioni hanno mostrato che la frequenza di GJB2 mutazione come causa di ipoacusia è molto variabile. Per esempio, tra famiglie segregante autosomica recessiva insufficienza uditiva non sindromica, GJB2varianti sono in correlazione causale con congenita deficit uditivo ereditario in circa il 25% delle famiglie palestinesi[Shahin et al 2002], almeno il 16% delle famiglie cinesi [Liu et al 2002] , circa il 22% della popolazione curda dell’Iran[Mahdieh et al 2004], e si stima che il 24% di Altaici dalla Siberia [Posukh et al 2005].
Diagnosi differenziale
Vedere Sordità ed Ereditaria Panoramica perdita dell’udito.
Autosomiche recessive sindromi con perdita dell’udito e:
- . La retinite pigmentosa tre tipi di sindrome di Usher sono riconosciuti; tutti sono ereditate in autosomica recessiva maniera. Usher sindrome tipo I è caratterizzata da congenita,, profonda perdita dell’udito neurosensoriale bilaterale; areflessia vestibolare; e adolescenti esordio retinite pigmentosa. A meno che non dotato di un impianto cocleare, gli individui con sindrome di Usher di tipo 1 in genere non sviluppano discorso.La retinite pigmentosa (RP), una progressiva, bilaterale, la degenerazione simmetrica delle funzioni di coni e bastoncelli della retina, si sviluppa in adolescenza, con conseguente campi visivi progressivamente ristretti e ridotta acuità visiva. La diagnosi di sindrome di Usher di tipo I è stabilita su basi cliniche utilizzando elettrofisiologico e test soggettivi di udito e funzione retinica. Varianti patogeniche nei geni a un minimo di nove diversi causa loci tipo sindrome di Usher I. Geni alle sei di questi loci – MYO7A (USH1B), USH1C, CDH23(USH1D), PCDH15 (USH1F), USH1G, e CIB2 (USH1J) – hanno stati identificati. sindrome di Usher di tipo II è caratterizzata da congenita, perdita bilaterale neurosensoriale dell’udito che è da lieve a moderata nelle frequenze basse e grave a profonda nelle frequenze più alte, le risposte vestibolari intatti, e retinite pigmentosa (RP) .Uno dei più importanti distinzioni cliniche tra Usher sindrome tipo I e sindrome di Usher di tipo II è che i bambini con sindrome tipo Usher I sono di solito in ritardo nel camminare fino all’età di 18 mesi a due anni a causa di coinvolgimento vestibolare, mentre i bambini con Usher sindrome tipo II di solito iniziano a camminare a circa all’età un anno. Tre geni sono noti per essere associati con la sindrome di Usher di tipo II: USH2A (pari al 80% dei casi), ADGRV1 (GPR98, VLGR1) (~ 15% dei casi), e DFNB31 (<5% dei casi). Un quarto locus è stato provvisoriamente mappato 15q. Sindrome di Usher tipo III è caratterizzato da postlinguale progressiva perdita dell’udito neurosensoriale, ad esordio tardivo RP, e variabili compromissione della funzione vestibolare. La mutazione di USH3 è causale. Gli individui più anziani con sindrome di Usher tipo III possono avere sordità profonda e disturbo vestibolare simile Usher sindrome tipo I.
- . Allargamento della tiroide sindrome Pendred viene diagnosticata in individui con: (1) problemi di udito che di solito è congenita e spesso grave a profonda, anche se lieve-moderata insufficienza progressiva dell’udito si verifica anche; (2) dilatazione bilaterale dell’acquedotto vestibolare (DVA, chiamato anche allargata acquedotto vestibolare o EVA) con o senza ipoplasia cocleare (DVA con ipoplasia cocleare è noto come Mondini malformazioni o displasia); e (3) o un test di scarico perclorato anormale o gozzo. Tiroide anomalia è variabile;modifiche goitrous non sono tipicamente presenti alla nascita, ma si sviluppano in pubertà precoce (40%) o in età adulta (60%). Inoltre, la funzione vestibolare è di solito anormale. Le analisi della sequenza di SLC26A4identifica varianti che provocano la malattia in circa il 50% dei colpiti individui da multiplex famiglie e il 20% degli individui provenienti da famiglie simplex. La trasmissione è autosomica recessiva.
- Difetti di conduzione cardiaca. Jervell e la sindrome di Lange-Nielsen (JLNS) comprende congenita profonda perdita bilaterale neurosensoriale dell’udito e lungo QTc, di solito superiore a 500 msec [Splawski et al 1997].Quest’ultimo è associato con tachiaritmie, che può condurre ad sincope o morte improvvisa. Oltre la metà dei bambini non trattati con JLNS morire prima dell’età di 15 anni. Il trattamento prevede l’uso di beta-bloccanti adrenergici, stimolatori cardiaci e defibrillatori impiantabili, nonché di evitare di farmaci che causano ulteriore prolungamento dell’intervallo QT e di attività noti per precipitare gli eventi sincopali. La diagnosi deve essere considerata in ogni bambino con congenita sordità neurosensoriale con i test DFNB1 negativo, soprattutto se il bambino ha una storia di sincope o sequestro o di una storia familiare di morte improvvisa prima dei 40 anni.Omozigosi per le varianti che provocano la malattia in uno KCNQ1 o KCNE1 è conferma. La trasmissione èautosomica recessiva.
Autosomica recessiva perdita di udito non sindromica senza identificabile GJB2 variante e con la progressione di perdita di udito:
- Con un acquedotto vestibolare dilatata sulla sottile taglio tomografia computerizzata (CT) delle ossa temporali suggerisce DFNB4 [Li et al 1998];
- Con una malformazione Mondini sulla sottile taglio CT delle ossa temporali suggerisce la sindrome di Pendred.Un test perclorato e test di genetica molecolare di SLC26A4 dovrebbero essere considerate [Everett et al 1997].
Altre cause di congenita grave perdita di udito da profonda dovrebbero essere considerati nei bambini che rappresentano casi singoli nella loro famiglia:
- Congenita da CMV (citomegalovirus), la causa più comune di congenita, perdita di udito non ereditaria
- Prematurità, basso peso alla nascita, a basso punteggio di Apgar, infezioni, e qualsiasi malattia che richiede cure in terapia intensiva neonatale
Gestione
Le valutazioni dopo la diagnosi iniziale
Per stabilire il grado di coinvolgimento e bisogni in un individuo con diagnosi di perdita di udito non sindromica e sordità (DFNB1), si consigliano le seguenti valutazioni:
- Valutazione completa di acuità uditiva utilizzando test appropriati all’età come il test ABR, uditivo risposta a regime di prova (ASSR), e audiometria tonale
- La valutazione oftalmologica per errori di rifrazione
Nota: È non possibile escludere retinite pigmentosa, una manifestazione dei tre tipi di sindrome di Usher, fino quasi alla fine della prima decade di vita. - Consultazione Genetica medica
Trattamento delle manifestazioni
I seguenti sono indicati:
- Raccordo con apparecchi acustici appropriati
- L’iscrizione in un programma educativo appropriato per i non udenti
- Esame di impianto cocleare (CI), un’opzione di abilitazione promettente per le persone con sordità profonda
- Il riconoscimento che, a differenza di molte condizioni cliniche, la gestione e il trattamento di gravi a profondecongenito sordità rientra in gran parte di competenza del benessere sociale e di sistemi d’istruzione, piuttosto che il sistema di assistenza medica [Smith et al 2005]
Sorveglianza
I seguenti sono adatti:
- Esame annuale da un medico familiare con deficit uditivo ereditario
- Ripetere audiometria per confermare la stabilità di perdita dell’udito
Agenti / Circostanze da evitare
Gli individui con perdita dell’udito dovrebbero evitare esposizioni ambientali noti per causare la perdita dell’udito. Il più importante tra questi per le persone con perdita dell’udito da lieve a moderata causata da mutazioni di GJB2 è evitare ripetute sovraesposizione a rumori forti.
Valutazione del Parenti a rischio
Chiarire lo status genetico di un bambino con una probabilità del 25% di avere DFNB1 dovrebbe essere considerato poco dopo la nascita in modo che un adeguato supporto e la gestione precoce possono essere forniti per il bambino e la famiglia.
Il test basato sul DNA può essere considerata solo se entrambe le varianti-sordità causa sono stati identificati in uncolpiti membro della famiglia.
Vedere consulenza genetica per le questioni legate alla sperimentazione di a rischio parenti per consulenza geneticascopi.
Terapie Sotto inchiesta
Cerca ClinicalTrials.gov per l’accesso alle informazioni su studi clinici per una vasta gamma di malattie e condizioni.Nota: Non ci possono essere gli studi clinici per questa condizione.
Consulenza genetica
La consulenza genetica è il processo di fornitura di individui e famiglie con informazioni sulla natura, l’ereditarietà, e le implicazioni di malattie genetiche per aiutarli a prendere decisioni personali e mediche informate. I seguenti sezione tratta la valutazione del rischio genetico e l’uso della storia familiare e test genetici per chiarire lo status genetico per i familiari. Questa sezione non è destinata a affrontare tutte le questioni personali, culturali, etiche o che gli individui possono affrontare o per sostituire la consultazione con una genetica professionali. -ED.
Modalità di eredità
Perdita di udito non sindromica e sordità (DFNB1) è trasmessa come autosomica recessiva maniera.
Rischio per i familiari
DFNB1 si verifica in persone che sono:
- Omozigoti o eterozigoti composti per GJB2 varianti;
- Eterozigoti composti per grande delezione di GJB2 5′-cis sequenze -regulatory che che includono una parte diGJB6.
I genitori di un probando
- I genitori sono eterozigoti obbligati e quindi portano una sola copia di una variante sordità causa.
- Gli eterozigoti sono asintomatici.
Fratelli e sorelle di un probando
- Al momento del concepimento, ogni sib ha una probabilità del 25% di essere sordo, una probabilità del 50% di essere un’udienza portante, e una probabilità del 25% di essere udito e non un vettore.
- Una volta che un sib a rischio è noto per essere udito, la possibilità della sua / suo essere un elemento portante è 2/3.
- Gli eterozigoti sono asintomatici.
Prole di un probando. Tutti i figli sono obligate portatori.
Altri membri della famiglia di un probando. Ogni sib di un eterozigote obbligato ha una probabilità del 50% di essere un elemento portante.
Carrier Detection
Carrier test di membri a rischio di famiglia è possibile se le varianti-sordità causa sono stati identificati in famiglia
Genetica Related Issues Counseling
Vedere Gestione, Valutazione di parenti a rischio per le informazioni sulla valutazione a rischio parenti ai fini della diagnosi precoce e del trattamento.
I seguenti punti sono degni di nota:
- La comunicazione con le persone che sono sordi richiede i servizi di un interprete qualificato.
- Le persone sorde possono visualizzare la sordità come una caratteristica distintiva e non come un handicap, perdita di valore, o patologie che richiedono una “cura” o “cura”, o da “impedito”. In effetti, avendo un bambino con la sordità può essere preferito avere un figlio con udito normale.
- Molte persone sorde sono interessati ad ottenere informazioni sulla causa della loro sordità, comprese le informazioni sui servizi sanitari, educativi e sociali, piuttosto che informazioni circa la prevenzione, la riproduzione o la pianificazione familiare. Come in tutta la consulenza genetica, è importante per il consulente di individuare, riconoscere e rispettare / della famiglia dell’individuo domande, dubbi e paure.
- L’uso di alcuni termini è preferito: probabilità o possibilità contro il rischio; non udenti e di udito contro udenti.Termini come “affetto”, “anormale” e “che causano malattie” dovrebbero essere evitati.
Pianificazione famigliare
- Il momento ottimale per la determinazione del rischio genetico, il chiarimento di vettore di stato, e la discussione della disponibilità di test prenatale è prima della gravidanza.
- È opportuno offrire una consulenza genetica per i giovani adulti non udenti o sono a rischio di essere portatori.
Banking DNA è la conservazione di DNA (tipicamente estratto da globuli bianchi) per un possibile uso futuro. Poiché è probabile che la metodologia di prova e la nostra comprensione dei geni, varianti alleliche, e le malattie migliorerà in futuro, occorre tenere in considerazione per bancaria DNA di affetti individui.
Test prenatale
Se le varianti alleliche-sordità causa sono stati identificati in un colpiti membro della famiglia, test prenatale per gravidanze a rischio può essere disponibile da un laboratorio clinico che offre sia il test per questa condizione / gene otest prenatale personalizzato.
Molte persone sorde sono interessati ad ottenere informazioni sulla eziologia della loro perdita di udito, piuttosto che informazioni sui rischi riproduttivi. È, quindi, importante accertare e affrontare le questioni e preoccupazioni della famiglia / individuo. “In contrasto con il modello medico che considera la sordità sia una condizione patologica, molte persone sorde non ritengono di essere portatori di handicap, ma si definiscono come facenti parte di un gruppo culturale distinta con una propria lingua, costumi e credenze. Strategie per efficace consulenza genetica per le persone non udenti includere il riconoscimento che la percezione del rischio è molto soggettiva e che alcuni individui sordi possono preferire di avere bambini sordi. ” (da Arnos et al [1991])
Le richieste di test prenatale per le condizioni come DFNB1 non sono comuni. Possono esistere differenze di prospettiva tra i professionisti medici e all’interno delle famiglie per quanto riguarda l’uso di test prenatali, in particolare se il test viene presa in considerazione ai fini della interruzione della gravidanza, piuttosto che la diagnosi precoce.Sebbene la maggior parte centri prenderebbero in considerazione decisioni su test prenatale per essere la scelta dei genitori, la discussione di questi problemi è appropriato.
Diagnosi genetica preimpianto (PGD) può essere un’opzione per alcune famiglie in cui sono state individuate le varianti-sordità causa.
Genetica molecolare
Le informazioni contenute in tabelle Genetica Molecolare e OMIM può differire da quello in altre parti del GeneReview: tavoli può contenere informazioni più recenti. – ED.
Tabella A.
Non sindromica, udito e Sordità, DFNB1: Geni e database
|
Locus Nome |
Gene |
Cromosoma Locus |
Proteina |
Locus specifico |
HGMD |
|
DFNB1 |
La homepage Connessina-sordità (GJB2) |
I dati sono compilati dai seguenti riferimenti standard: gene da HGNC; cromosoma locus, nome luogo, regione critica, complementazione gruppo da OMIM; proteine da UniProt. Per una descrizione di database (Locus specifico, HGMD) a cui vengono forniti i link, clicca qui.
Tabella B.
OMIM Voci per non sindromica, udito e sordità, DFNB1 (Visualizza tutto in OMIM)
|
GAP JUNCTION PROTEINA, BETA-2; GJB2 |
|
|
SORDITA ‘, AUTOSOMICA 1A RECESSIVA; DFNB1A |
Struttura genica. La maggior parte connexine hanno un’architettura comune, con l’intera regione codificante contenuta in un unico grande esone separato dalla regione 5′-non tradotta da un introne di dimensioni variabili. La sequenza codifica di GJB2 (esone 2) 681 paia di basi (compreso l’arresto codone) e si traduce in una proteina di 226-amino. Per un riepilogo dettagliato delle gene informazioni e proteine, vedi tabella A, Gene.
Varianti alleliche patogeni. Vedi Tabella 2. Numerosi diverso-sordità causando varianti di GJB2 che si traducono inautosomica recessiva perdita di udito non sindromica sono elencati nella Home page Connessina-sordità. La variante più comune nelle persone di discendenza europea settentrionale è la variante c.35delG. Questa variante è stata riportata anche in individui di lingua araba, Beduino, indiana, pakistana ed etnia. Sulla base di stretti collegamenti con singlenucleotide polimorfismi (SNP), una variante fondatore provenienti da sud Europa circa 10.000 anni fa, è stato previsto[Van Laer et al 2001]. Coerentemente con questa previsione è un c.35delG sordità gradiente nord-ovest a sud-est attraverso i paesi del Golfo Persico [Najmabadi et al 2005] e un c.35delG pendenza sordità sud-a-est in Europa[Gasparini et al 2000, Lucotte & Mercier 2001, Rothrock et al 2003].
Lo spettro di GJB2 varianti patogeniche diverge sostanzialmente tra le popolazioni, come evidenziato dagli specifici pregiudizi etnici per le varianti più comuni. Come accennato in precedenza, il c.35delG allele è comune tra individui di origine nord-europea, con un tasso di vettore di 2% al 4% [Estivill et al 1998, Green et al 1999]; considerando c.235delC è più comune nella popolazione giapponese (portatore rate: 1% al 2%) [Abe et al 2000, Kudo et al 2000];c.167delT è più comune nel ashkenazi popolazione (tasso vettore: 7,5%) [Morell et al 1998]; e p.Val37Ile è più comune in Thailandia (portatore rate: 11,6%) [Hwa et al 2003]. (Per ulteriori informazioni, vedere la tabella A).
La variante p.Met34Thr è stato descritto prima come autosomica dominante variante patogena [Kelsell et al 1997], in linea con lo studio di White et al [1998] in cui è stato segnalato per avere un effetto dominante negativo over wild-type connessina 26 in Xenopus ovociti. Questo risultato, tuttavia, è stato successivamente attribuito ad un manufatto in livelli di espressione di mRNA mutant- e wild-type che non sono stati controllati nel sistema esogeno [Skerrett et al 2004].
C’è una forte evidenza per classificare la p.Met34Thr allele come autosomica recessiva variante patogena [Wilcox et al2000, Houseman et al 2001, Kenneson et al 2002, Wu et al 2002]. Supponendo che la variante p.Met34Thr di essere un benigno polimorfismo, le persone sorde che sono eterozigoti composti per [c.35delG] + [p.Met34Thr] potrebbero essere portatori di una sola GJB2 variante (c.35delG) e la loro perdita di udito devono essere provocati da altre varianti non identificati presso la DFNB1 locus o di altri geni. A causa della grande variabilità fenotipica visto con deficit uditivo genetico, un simile grado di variabilità in perdita dell’udito ci si aspetterebbe in questi individui. Tuttavia uno studio su 38 soggetti che erano eterozigoti composti per [c.35delG] + [p.Met34Thr] ha dimostrato che tutti avevano la perdita dell’udito da lieve a moderata con un PTA mediana 0.5,1,2kHz di 34 dB [Snoeckx et al 2005 ]. I 16 soggetti omozigoti per p.Met34Thr avuto un ancora più basso mediano PTA 0.5,1,2khz valore (30 dB) [Snoeckx et al 2005].
La variante p.Val37Ile è stato anche segnalato come non patogeno [Kelley et al 1998, Kudo et al 2000, Hwa et al 2003,Wattanasirichaigoon et al 2004]; tuttavia, Snoeckx et al [2005] hanno documentato un’associazione di questa variante con perdita dell’udito lieve in nove su dieci combinazioni genotipiche. Questo risultato è in linea con altri studi del allele[Abe et al 2000, Wilcox et al 2000, Kenna et al 2001, Lin et al 2001, Marlin et al 2001].
Tabella 2.
Selezionati GJB2 patogeni Varianti
|
DNA Nucleotide Change |
Proteine Amino Acid Change |
Sequenze di riferimento |
|
c.101T> C |
p.Met34Thr 2 |
|
|
c.109G> A |
p.Val37Ile 2 |
|
|
c.35delG |
p.Gly12ValfsTer1 |
|
|
c.35G> T |
p.Gly12Val |
|
|
G.-3179G> A 3 |
– |
|
|
c.56G> C |
p.Ser19Thr |
|
|
c.167delT |
p.Leu56ArgfsTer26 |
|
|
c.235delC |
p.Leu79CysfsTer3 |
|
|
c.231G> A |
p.Trp77Arg |
|
|
c.269T> C |
p.Leu90Pro |
|
|
c.339T> G |
p.Ser113Arg |
|
|
c.358_360delGAG |
p.Glu120del |
|
|
c.427C> T |
p.Arg143Trp |
|
|
c.487A> G |
p.Met163Val |
|
|
c.551G> C |
p.Arg184Pro |
Nota sulla classificazione variante: Varianti elencate nella tabella sono stati forniti dagli autori. GeneReviews personale non ha verificato in modo indipendente la classificazione delle varianti.
Nota sulla nomenclatura: GeneReviews segue le convenzioni di denominazione standard del Human Genome Variation Society (www .hgvs.org). Vedere di riferimento rapido per una spiegazione di nomenclatura.
1.Designazione Variante che non è conforme alle convenzioni di denominazione corrente
2.p.Met34Thr e p.Val37Ile sono associati con udito normale o la perdita dell’udito lieve. Si veda la discussione in varianti allelichepatogeni.
3.IVS1 + 1G> A è -3179 nucleotidi dall’inizio della esone 2 della sequenza genomica (Riferimento Sequenza NC_000013 0,9)
Le varianti patogeni associati DFNB1 sono grandi delezioni che includono gran GJB6 e una grande porzione della regione upstream. Nonsense o missenso varianti di GJB6 che potrebbero essere rilevati da analisi di sequenza non sono stati associati con DFNB1. Le eliminazioni si ritiene di influenzare la trascrizione di GJB2 presumibilmente eliminando una cis elemento -regulatory. (Per ulteriori informazioni, vedere la tabella A).
Il Δ GJB6 -D13S1830 eliminazione è il più comune GJB6 variante associata con DFNB1. La soppressione è più frequente in Spagna, Francia, Regno Unito, Israele e Brasile (origine portoghese), dove rappresenta il 5,9% e il 8,3% di tutte le DFNB1 alleli. In uno studio, per esempio, il 67% degli individui sordi spagnoli con quello identificato GJB2variante effettuata questa soppressione [Wu et al 2002, Stevenson et al 2003]. La sua frequenza è più bassa in Belgio e Australia (1,3% -1,4%), e non è stato trovato tra sordi italiani GJB2 eterozigoti. Negli Stati Uniti, la sua frequenza è del 1,6% al 4,0% [Del Castillo et al 2003].
I Δ GJB6 conti -D13S1854 variante per circa il 25% dei sordi GJB2 eterozigoti che sono rimasti irrisolti dopo lo screening per Δ GJB6 -D13S1830 in Spagna; esso rappresenta il 22,2% nel Regno Unito, 6,3% in Brasile, e l’1,9% nel Nord Italia. Questa eliminazione non è stato trovato in sordi GJB2 eterozigoti provenienti da Francia, Belgio, Israele, l’Autorità Palestinese, gli Stati Uniti, o in Australia. Le analisi di aplotipo ha rivelato uno dei fondatori comune per la variante in Spagna, Italia e Regno Unito [del Castillo et al 2005].
Una terza eliminazione è stata identificata in una grande famiglia tedesco-americano, in cui 15 persone sono recessiva ereditaria congenita, grave a profonda sordità neurosensoriale non sindromica. Undici di questi 15 persone sono omozigoti per la GJB2 variante 35delG; tuttavia, i restanti quattro portano una sola variante di 35delG. Ridotta espressione di entrambi GJB2 e GJB6 mRNA dal allele portato in trans con la variante che il cuscinetto 35delG in queste quattro persone era coerente con la presenza di una delezione upstream, confermata mediante array comparativegenomica ibridizzazione. Questa eliminazione – del (CHR13: 19,837,344-19,968,698) – è 131,4 kb. Il suo punto di rottura prossimale si trova più di 100 kb a monte dei siti di inizio trascrizionale di GJB2 e GJB6 e segrega come completamente penetranti allele-DFNB1 causando in questa famiglia [Wilch et al 2010].
Tabella 3.
cis -Regulatory 5 ‘GJB 2 patogena Varianti
|
DNA Nucleotide Modifica 1 |
Proteine Amino Acid Change |
Sequenze di riferimento |
|
Δ GJB6 -D13S1830 |
– |
|
|
Δ GJB6 -D13S1854 |
– |
|
|
del (CHR13: 19,837,344-19,968,698) |
– |
Umano genoma sequenza di riferimento (NCBI Costruire 36.1 oversione UCSC hg18) |
Nota sulla classificazione variante: Varianti elencate nella tabella sono stati forniti dagli autori. GeneReviews personale non ha verificato in modo indipendente la classificazione delle varianti.
Nota sulla nomenclatura: GeneReviews segue le convenzioni di denominazione standard del Human Genome Variation Society (www .hgvs.org). Vedere di riferimento rapido per una spiegazione di nomenclatura.
1.
Le denominazioni sono varianti colloquiali di uso comune e non sono conformi alle convenzioni di denominazione corrente.
Normale gene prodotto. Connexin 26 è una proteina beta-2 gap giunzione composta di 226 amminoacidi. Connessine aggregano in gruppi di sei intorno ad una centrale di 2,3 nm pori per formare una connessone. Connexons da celle adiacenti in modo covalente legame formando un canale tra le cellule. Grandi aggregazioni di connexons chiamati placche sono i costituenti di giunzioni. Giunzioni consentono scambi intercellulari diretto di ioni e molecole attraverso i loro pori acquosi centrali. Ruoli postulate includono la rapida propagazione di segnali elettrici e la sincronizzazione di attività in tessuti eccitabili e lo scambio di metaboliti e molecole segnale nei tessuti non-eccitabili.
Una proteina connessina contiene due extracellulare (E1-E2), quattro transmembrana (M1-M4), e tre domini citoplasmatici. Ogni dominio extracellulare ha tre residui di cisteina con almeno un legame disolfuro adesione E1 ed E2 loop. La presunta importanza di questi sei cisteine si può dedurre da connessina 32 esperimenti in cui qualsiasi cisteinamutazione blocca completamente lo sviluppo di conduttanze gap-giunzione tra Xenopus coppie ovociti. Il dominio transmembrana terzo (M3) è anfipatico e le linee della parete putativo del canale connessone intercellulare. Se i connexons fornite da ciascuna cella sono composti della stessa connessina, il canale è omotipica; se ogni connessone è formato da un connessina diversa, è heterotypic. Con l’eccezione di connessina 26, tutte le connessine sono fosfoproteine. Connessina 26 forme combinazioni funzionali con sé, connessina 32, connessina 46, e connessina 50.
Anormale gene prodotto. Canali Gap di giunzione sono permeabili a ioni e piccole metaboliti con masse molecolari relative fino a circa 1,2 kd [Harris & Bevans 2001]. Le differenze nei meccanismi di selettività e di gating ionici tra giunzioni riflettono l’esistenza di più di 20 diversi connessina isoforme negli esseri umani. Solo pochi GJB2 varianti patogeni sono stati testati in sistemi di espressione ricombinanti, con la maggior perdita proiezione di funzione come risultato di smistamento alterata (p.Gly12Val, p.Ser19Thr, c.35delG, p.Leu90Pro), incapacità di indurre la formazione di omotipica canali gap junction (p.Val37Ile, p.Trp77Arg, p.Ser113Arg, p.Glu120del, p.Met163Val, p.Arg184Pro e c.235delC), o interferenza con traduzione (p.Arg184Pro) [Snoeckx et al 2005].
Chapter Notes
Acknowledgments
Supported in part by grant RO1-DC02842 from the NIDCD (RJHS)
Author History
Daryl A Scott, MD, PhD; University of Iowa (1998-2001)
Val C Sheffield, MD, PhD; University of Iowa (1998-2001)
Richard JH Smith, MD (1998-present)
Guy Van Camp, PhD (1998-present)
Revision History
- 2 January 2014 (me) Comprehensive update posted live
- 14 July 2011 (me) Comprehensive update posted live
- 11 July 2008 (me) Comprehensive update posted to live Web site
- 21 December 2005 (me) Comprehensive update posted to live Web site
- 14 March 2005 (rjs) Revision: information on GJB6 deletions
- 15 July 2004 (rjs) Revision: use of an interpreter
- 27 October 2003 (me) Comprehensive update posted to live Web site
- 24 April 2001 (me) Comprehensive update posted to live Web site
- 28 September 1998 (pb) Review posted to live Web site
- 4 April 1998 (rjs) Original submission
Copyright © 1993-2016, University of Washington, Seattle. All rights reserved.
For more information, see the GeneReviews Copyright Notice and Usage Disclaimer.
For questions regarding permissions: ude.wu@tssamda.
Bookshelf ID: NBK1272PMID: 20301449
Quattro geni delle connexine, espressi nell’orecchio interno, possono essere implicati in ipoacusie isolate o sindromiche:
• GJB2(connexina26.inl3qll.q12)
• GJB6 (connexina 3Oinl3qll.q12)
• GJB3 (connexina 32. in Xql3.l)
• G.JB i (connexina 31. in lp34).
GJB2 è responsabile della forma principale (50% dei casi) di ipoacusia genetica, DFNB 1: più recentemente è stato dimostrato che una ipoacusia DFNB 1 può essere dovuta anche all’associazione di una delezione di GJB6. situato nello stesso locus del GJB2. con una mutazione di GJB2.
Si riteneva inizialmente che le connexine 26 e 30 fossero coinvolte nel ricircolo del potassio bell’endolinfa e che l’ipoacusia potesse essere dovuta a un’alterazione della concentrazione di K endolinfatico. La patogenesi è meglio conosciuta grazie a modelli animali di delezione parziale o totale dei geni GJB2 e GJB6 che dimostrano che l’orecchio interno si sviluppa normalmente e che la morte cellulare sopraggiunge verso il l4 giorno postnatale nel topo. Inizialmente sono interessate le cellule di sostegno e poi le cellule ciliate. La concentrazione di potassio nell’endolinfa rimane normale tino a questa data. facendo avanzare l’ipotesi di una tossicità scatenata dalla risposta delle cellule ciliate interne alla stimolazione sonora. per esempio a causa di un mancato ricircolo del glutammato ,
Gene della connexina 26 (CX26 o GJB2)
La mutazione di GJB2 è responsabile sia della forma più comune di ipoacusia del bambino, quella autosomica recessiva DFNB 1, sia di una forma rara di ipoacusia autosomica dominante, la DFNA35. Altre mutazioni di questo gene rendono conto anche di tre sindromi rare che associano ipoacusia e interessamento cutaneo, la sindrome di Vohwinkel, l’ipoacusia con cheratosi palmoplantare e la sindrome Keratosis Ittiosi Deafness (KID)6 7.
Nella ipoacusia DFNB i, i soggetti sordi portano delle mutazioni sui due alleli del gene, mentre i soggetti eterozigoti sono normoudenti. L’ipoacusia è congenita e stabile nella maggioranza dei casi (un’evolutività è osservata in meno del 20% dei casi, in genere lieve) ed è più spesso profonda e pantonale8.
La TC delle rocche petrose e le prove vestibolari caloriche sono normali.
Le mutazioni individuate sono allo stato oltre 60 ma la mutazione che predomina nei paesi occidentali è la 35delG, dovuta alla delezione di una guanina in posizione 35 nella parte codificante del gene e che determina la formazione di una proteina tronca. Questa mutazione rappresenta il 55 66% degli alleli mutati in Italia, il 46% negli Stati Uniti9. I portatori eterozigoti (normoudenti) di questa mutazione sono molto frequenti nella popolazione generale. tra il 2,5 e il 4% della popolazione in Italia, il 2% negli Stati Uniti10 Il
Le mutazioni “inattivanti” che portano auna proteina tronca sono associate ad ipoacusia più grave delle mutazioni “non inattivanti’5. In particolare la mutazione frequente L9OP associata a 35 del G sull’altro allele (genotipo L9OP/35delG) è più comunemente associata ad una ipoacusia medio-lieve rispetto al genotipo 35delG!35delG.
Un’altra mutazione frequente. M34T. non risulta patogena nella maggior parte dei casi: sono stati segnalati molti soggetti 35delG/M34T normoudenti’.
Nel 5% dei casi si riscontra inoltre una delezione del gene della connexina 30, situato nell’intervallo del locus DFNB 1, in associazione con una mutazione del GJB2 sull’altro allele,
Le mutazioni del gene della connexina 26 sono coinvolte anche nella forma di ipoacusia autosomica dominante DFNA3. Chiaramente patogene sono le mutazioni W44C e C202F’.
Le caratteristiche dell’ipoacusia sono molto diverse a seconda delle mutazioni: per esempio. per W44C l’ipoacusia è prelinguale, da media a profonda e colpisce tutte le frequenze. mentre per C202F compare tra 1O e 20 anni. colpisce inizialmente le alte frequenze ed evolve lentamente per divenire media-lieve a 50 anni.
Altri geni delle connexine
Altri geni delle connexine possono essere responsabili di una ipoacusia isolata. autosomica recessiva e/o dominante: GJB6 (coliliexina 30)
• GJB3 (connexina 31).
Il gene GJB6 (connexina 30) è particolarmente importante: molto simile al gene della connexina 26, una delezione di un singolo allele di questo gene (GJB6- Dl3Sl83Odel) è frequentemente riscontrata in associazione con una mutazione eterozigote del gene della connexina 26 in soggetti sordicongeniti’4.
Sono invece implicati in ipoacusie sindromiche:
• GJB i (connexina 32), responsabile di una forma della sindrome di Charcot-MarieTooth’ 5
• GJA1 (connexina 43), coinvolto nella displasia oculo-dento-digitale in cui si riscontra una ipoacusia trasmissiva16,
La diagnosi molecolare di mutazioni del GJB2!GJB6 è la prima ad essere proposta al termine della valutazione eziologica in presenza di una ipoacusia non sindromica prelinguale se la diagnostica per immagini della rocca petrosa è normale o se non ha potuto essere realizzata a causa della giovane età del paziente.
Nell’adulto bisogna chiedere queste indagini molecolari quando l’ipoacusia risale all’infanzia
Il gene connessina 26 è il gene maggiormente responsabile dei casi di sordità recessiva. Nel 1997, il lavoro svolto a Londra da D.P. Kelsell, ha dimostrato che le mutazioni della connessina 26 sono responsabili di circa il 50% dei casi di sordità recessiva nel bacino del Mediterraneo. Questa scoperta è stata molto importante, poiché il lavoro che ne è seguito ha dimostrato che le mutazioni della connessina 26 sono presenti anche nella maggior parte delle altre popolazioni: 30% in Nord Europa, 40% negli Stati Uniti, 22% in Brasile, 25% in Estremo Oriente, 17% in Ghana e 15% in Australia. Queste percentuali dimostrano che lo studio della connessina 26 ha delle implicazioni rilevanti nella diagnosi molecolare e nella consulenza ai pazienti.
La maggior parte delle mutazioni della connessina 26 sono trasmesse tramite modalità di eredità recessiva, anche se in alcuni casi sono state individuate rare mutazioni causa di sordità dominante o di forme sindromiche di sordità associate a malattie della pelle. A tutt’oggi, sono state rilevate più di 80 diverse mutazioni della connessina 26, elencate nella homepage sulla sordità-Cx26 all’indirizzo
http://www.crg.es/deafness/cx26mut.php.
Nella modalità di eredità recessiva, i genitori sono portatori sani con due copie di ogni gene: uno è mutato e l’altro no. Poiché è necessaria la presenza di due copie mutate per causare una perdita uditiva, quando avranno dei bambini, essi avranno un 25% di probabilità che i bambini saranno sordi, un 50% di probabilità che i bambini avranno un udito normale ma saranno portatori di una copia mutata e di una copia normale, e un 25% di probabilità che i bambini avranno un udito normale e non saranno portatori di alcuna copia mutata.
Nella modalità di eredità dominante non ci sono portatori sani. Pertanto è sufficiente una copia mutata per causare la perdita dell’udito. Per esempio, un genitore con menomazione uditiva ha solo una copia mutata e l’altra copia è normale. Questa persona sposa un partner con udito normale. Lui o lei hanno un 50% di probabilità di avere un bambino con menomazione uditiva e un 50% di probabilità di avere un bambino con l’udito normale.
Fra le popolazioni del Mediterraneo europeo, la mutazione più frequente di questo gene è 35delG che è responsabile dell’80% delle mutazioni. Le frequenze del portatore sano variano da 1/35 nell’Europa meridionale a 1/79 nell’Europa settentrionale. Ciò significa che almeno il 3% della popolazione dell’area del Mediterraneo è portatrice sana.
La correlazione tra il tipo di mutazione che si verifica in questo gene e il grado/gravità di perdita d’udito non è chiara. Tuttavia, parlando in generale, sembra che le persone con 35delG in entrambi le copie del gene abbiano una perdita di udito più grave (sordità grave/profonda) rispetto ai pazienti
![]() con una mutazione 35delG e una mutazione non-35delG. Le persone che presentano due mutazioni non-35delG hanno una perdita dell’udito ancora meno grave (sordità moderata).
con una mutazione 35delG e una mutazione non-35delG. Le persone che presentano due mutazioni non-35delG hanno una perdita dell’udito ancora meno grave (sordità moderata).
In altre popolazioni sono presenti con altissima frequenza le seguenti mutazioni: 167delT fra gli Ebrei Ashkenazi, 235delC fra le popolazioni dell’Asia Orientale (Giapponesi, Cinesi e Coreani), R143W in Ghana e W24X fra le popolazioni dell’India e della Romania (gitani).
Il gene connessina 26 (GJB2 – Gap Junction Beta protein 2) codifica una proteina che è una delle sei subunità identiche che si uniscono per formare un connessone, la metà di un canale. Infatti, due connessoni in cellule opposte formano un canale funzionale, Gap Junction (giunzione di connessione), che connette due cellule adiacenti e consente il passaggio di sostanze chimiche come lo ione di potassio.
La connessina 26 è espressa nelle cellule di supporto dell’organo di Corti e delle cellule epiteliali adiacenti, nei fibrociti del lembo spirale e della prominenza spirale e nelle cellule basali e intermedie della stria vascularis. La sua funzione non è ancora totalmente conosciuta, ma si ipotizza che sia coinvolta nel riciclaggio nell’endolinfa degli ioni potassio che entrano nelle cellule ciliate in seguito alla trasduzione sonora.
L’importanza delle altre connessine.
Nella famiglia di proteine connessina ci sono due altre connessine, la 30 e la 31, che sono simili alla 26 e che sono causa di sordità. La Connessina 31 sembra essere responsabile sia della sordità dominante che di quella recessiva, ma per provare questa teoria sono necessarie ulteriori ricerche. Perciò, possiamo costatare che differenti mutazioni nello stesso gene possono provocare differenti modalità di ereditarietà della sordità.
Il gene connessina 30 è posizionato molto vicino al gene connessina 26. Poiché un’alta percentuale di pazienti con perdita dell’udito (10%-42% in diverse popolazioni) portano una sola copia mutante in connessina 26, gli scienziati hanno ipotizzato che la seconda copia mutante potrebbe trovarsi in un gene vicino a connessina 26. Essi hanno riscontrato che nel 50% delle persone non udenti che avevano solo una mutazione in connessina 26 mancava anche un frammento di 309 kb dell’altro cromosoma, che eliminava una delle due copie di connessina 30. Questa mutazione è stata chiamata delezione GJB6-D13S1830.
E’ stata identificata anche un’altra delezione di 232 kb (chiamata GJB6-D13S1854) che riguarda il 25% dei casi rimasti irrisolti in seguito allo screening per la delezione di 309 kb. Il resto delle mutazioni negli eterozigoti affetti da connessina 26 (10-30% dei casi) resta non identificato. La scoperta che nell’orecchio le subunità connessina 26 e 30 si combinano per formare giunzioni di connessione funzionali, spiega il ruolo importante della connessina 30. Inoltre, in poche famiglie, la connessina 30 è responsabile di una forma dominante di sordità.
La connessina 43 (GJA1) che è un altro gene appartenente alla famiglia delle connessine, è stata dimostrata essere responsabile di displasia oculo-dento-digitale (ODDD), una sindrome autosomica dominante associata, in alcuni pazienti, a perdita uditiva conduttiva (anomalia nell’orecchio esterno o medio).
Invece, si sa ormai che le mutazioni in connessina 45 non provocano sordità sensoriale sindromica, come riportato nel 2001.
Le mutazioni a carico del gene GJB2 che codifica per la connessina 26 sono ritenute responsabili di più del 50% di queste forme di sordità, almeno a livello del bacino del Mediterraneo (4). Le percentuali nel resto del mondo sono varie: 30% nel Nord Europa, 40% negli Stati Uniti, 22% in Brasile, 25% in Estremo Oriente, 17% in Ghana e 15% in Australia (5).
Il locus contenente il gene GJB2 è localizzato sul cromosoma 13 (6). La prima mutazione scoperta a carico di tale gene fu individuata per in famiglie consanguinee della Tunisia (7) ma un importante contributo fu dato, in seguito, da studi effettuati in 19 famiglie caucasiche (8).
Mutazioni nella cx 26 possono anche essere, in rari casi, cause di sordità autosomiche dominanti: Sono state individuate nove mutazioni responsabili di sordità dominanti tra cui: M34T, W44C, W44S, R75W, D66H (5,12).
Anche se la maggior parte delle sordità ereditarie è imputata ad una mutazione del gene GJB2 in una condizione di omozigosi, sono stati trovati casi in cui non erano presenti mutazioni della cx26 o si presentava una condizione di eterozigosi quindi la mutazione di un solo allele. Studi su questi casi hanno suggerito la presenza di altri geni coinvolti nella sordità e collocati sullo stesso punto nel cromosoma. Questa ipotesi è stata confermata dal ritrovamento di due delezioni che non riguardano il gene GJB2 ma interessano il gene GJB6 che codifica per la connessina 30, localizzato nello stesso locus di GJB2. Queste delezioni chiamate del(GJB6-D13S1830) e del(GJB6-D13S1854), sono state trovate in soggetti omozigoti o in doppia eterozigoti con la mutazione di GJB2 (13,14) .
Studi hanno dimostrato che la prima delezione (del(GJB6-D13S1830)), è molto frequente in Spagna, Francia, Inghilterra, Israele e Brasile, è meno frequente negli USA, Belgio e Australia e molto rara nel Sud Italia (15). La seconda delezione, identificata più recentemente, anch’essa molto frequente in Spagna, è stata identificata solo nel Nord Italia.
Inoltre è stato riscontrato che mutazioni di GJB6 possono essere responsabili di sordità autosomiche dominanti (16).
Anche altre mutazioni nei geni che codificano per le restanti connessine presenti in percentuali minori nell’orecchio interno possono essere coinvolte nei disturbi uditivi e causare sordità dominanti, recessive o partecipare nelle forme sindromiche. I geni interessati sono: GJB1 che codifica per la connessina 32, una mutazione in questo gene può causare la forma X-linked Charcot-Marie-Thoot di tipo 1 (17); GJB3 che codifica per la connessina 31:
una mutazione in questo gene può portare sia sordità che disturbi cutanei (18), a seconda di dove è collocata la mutazione.
III APPROFONDIMENTO
Non sindromica, udito e sordità, DFNA3
Richard JH Smith, MD, Paul T Ranum, BA, e Guy Van Camp, PhD.
Distacco iniziale: 28 settembre 1998; Ultimo aggiornamento: 12 Giugno 2014.
Sommario
Caratteristiche cliniche.
Perdita dell’udito non sindromica e sordità, DFNA3, è caratterizzata da pre- o postlinguale, da lieve a profonda, ad alta frequenza insufficienza uditiva neurosensoriale progressiva. Gli individui affetti hanno altre constatazioni mediche associate. La maggior parte delle persone con diagnosi di DFNA3 avere un genitore sordo; lastoria della famiglia è raramente negativo.
Diagnosi / testing.
DFNA3 è causato da presenza di una dominante -negativa variante patogena in GJB2 o GJB6alterazione della proteina connessina 26 (Cx26) o connessina 30 (Cx30), rispettivamente, o. La diagnosi dipende test genetici molecolari per identificare una variante patogena in entrambi gene.
Gestione.
Trattamento delle manifestazioni: Raccordo con gli apparecchi acustici e di adeguati programmi educativi.L’impianto cocleare può essere eseguita per le persone con sordità profonda.
Sorveglianza: audiogramma semestrale dopo la diagnosi iniziale.
Agenti / circostanze da evitare: esposizioni ambientali noti per causare la perdita dell’udito, come forti rumori ripetuti.
La valutazione dei parenti a rischio: i test genetici molecolari per a rischio parenti di individui con una nota variante patogena; audiometria tonale pura per i membri a rischio della famiglia quando i test genetici molecolari non è disponibile.
Consulenza genetica.
DFNA3 è ereditata come autosomica dominante maniera. Prole di un affetto individuo hanno una probabilità del 50% di ereditare il alterata del gene. Test prenatale per gravidanze a rischio è possibile se la variante patogena nella famiglia è nota.
Diagnosi
La diagnosi di perdita di udito non sindromica e sordità, DFNA3 deve essere sospettata in individui con il seguente:
- Pre- o postlinguale, da lieve a profonda, progressivo deterioramento dell’udito neurosensoriale [Denoyelle et al 2002] Nota: (1) Audizione si misura in decibel (dB). La soglia o 0 dB contrassegno per ogni frequenza si riferisce al livello al quale normali giovani adulti percepiscono un Tone Burst 50% del tempo. Audizione è considerato normale se le soglie di un individuo sono a meno di 15 dB di soglie normali. (2) La gravità della perdita dell’udito è classificato come lieve (26-40 dB), moderata (41-55 dB), moderatamente grave (56-70 dB), grave (71-90dB), o profonda (> 90dB). La frequenza di perdita dell’udito è designato come basso (<500 Hz), medio (501-2000Hz), o alto (> 2000 Hz) (vedi ereditaria, udito e sordità Panoramica).
- Nessun risultati sistemici correlati identificati da anamnesi e l’esame obiettivo
- Una storia familiare di perdita di udito non sindromica coerente con autosomica dominante ereditarietà
La diagnosi di perdita di udito non sindromica e sordità, DFNA3 è stabilito in un probando dal rilevamento di una variante patogena in GJB2 o GJB6. GJB2, che codifica connessina 26, e GJB6, che codifica per la connessina 30, sono gli unici due geni nei quali varianti patogeniche sono noti per causare sordità al DFNA3 locus (vedi tabella 1).GJB2 è condivisa da un locus di sovrapposizione, DFNB1, che è associato con autosomica recessiva perdita di udito non sindromica. Varianti in GJB2 possono causare sia autosomica dominante recessiva perdita DFNA3 associata o autosomica DFNB1 associata non sindromica udito e la sordità. La modalità di trasmissione è in definitiva determinato da come gli impatti variante l’espressione e la funzione della proteina connessina 26.
La strategia di test genetici preferito è l’uso di un multi gene pannello che include GJB2 e GJB6 nonché un certo numero di geni in cui mutazione provoca disturbi associati alla perdita (descritto nel sentire diagnosi differenziale).Nota: I geni inseriti e metodi utilizzati in multi-gene pannelli variano da laboratorio e nel tempo. Pannelli Multi-gene sono state sviluppate per schermare gli esoni codificanti di tutti i geni implicati nella perdita di udito non sindromica.
Una strategia alternativa test genetici è singolo gene testing:
- Il primo passo per la diagnosi genetica di DFNA3 è l’analisi di sequenza di GJB2 esone 2.
- Se non varianti patogeniche sono identificati in GJB2, prendere in considerazione la sequenza di GJB6, con l’avvertenza che solo due riportato DFNA3 provocano varianti patogeniche di GBJ6 sono stati segnalati.
- Se non viene trovata alcuna variante patogena, analisi delezione / duplicazione può essere effettuata sia per GJB2e GJB6.
È importante notare che un intero multi gene pannello può spesso essere completata per meno spese di Sanger sequenziamento di un grande gene. Pannelli Multi-gene sono agnostico alla audioprofile e quindi non necessitano di ‘conoscenza di esperti’ per prevedere una causa genetica probabile di perdita dell’udito per il sequenziamento Sanger.
Vedere rilevante ACMG ACT Foglio e ACMGalgoritmo.
Tabella 1.
Sintesi di ricerca di genetica molecolare Utilizzato in non sindromica, udito e sordità, DFNA3
|
Gene 1 |
Percentuale di DFNA3 Attribuito a mutazioni di questo gene |
Metodo di prova |
|
GJB2 |
> 90% |
Le analisi della sequenza 2, 3 / scansione mutazione 4 |
|
Cancellazione / duplicazione analisi 6, 7 |
||
|
GJB6 |
<10% |
Sequenza analisi 2, 4, 8 |
|
Cancellazione / duplicazione analisi 6, 7 |
1.
Vedi Tabella Geni A. e database per cromosoma locus e proteine. Vedere Genetica Molecolare per informazioni su varianti alleliche.
2.
Le analisi della sequenza rileva le varianti che sono benigni, probabilmente benigno, di significato incerto, probabilmente patogeni o patogeni. Varianti patogeniche possono includere piccole intragenica delezioni / inserzioni e missense, nonsense, e varianti sito di splicing; tipicamente, esone o all’in- grosso gene delezioni / duplicazioni non vengono rilevati. Per problemi da considerare nell’interpretazione di analisi di sequenza risultati, fare clic qui.
3.
Le analisi della sequenza di GJB2 identifica il 100% di varianti patogeni, tra cui p.Trp44Cys, p.Trp44Ser, p.Asp46Asn, p.Thr55Asn, p.Pro58Ala, p.Arg75Gln, p.Arg75Trp, p.Arg143Gln, p.Met163Leu, pag. Asp179Asn, p.Arg184Gln, e p.Cys202Phe, i dieci varianti patogeniche segnalati a segregare in persone con DFNA3 [Denoyelle et al 1998, Morle et al 2000, Hamelmann et al 2001, Janecke et al 2001, Löffler et al 2001, Marziano et al 2003, Primignani et al 2003, Feldmann et al 2005, Melchionda et al 2005, Piazza et al2005, Primignani et al 2007, Matos et al 2008, Bazazzadegan et al 2011].
4.
Le analisi della sequenza e scansione mutazione dell’intero gene possono avere frequenze rilevamento mutazione simili; tuttavia, i tassi di rilevamento di mutazione per la scansione mutazione possono variare notevolmente tra laboratori a seconda del protocollo specifico utilizzato.
5.
Nota: le varianti patogeni inclusi in un pannello può variare da laboratorio.
6.
Test che identifica esone o all’in- grosso gene delezioni / duplicazioni non rilevabili mediante analisi della sequenza di codifica e regioni fiancheggianti intronic di genomica DNA. Incluso nel varietà di metodi che possono essere utilizzati sono: la PCRquantitativa, a lungo raggio PCR, legatura-dipendente multipla della sonda amplificazione (MLPA), e cromosomica microarray(CMA) che include questo / gene del cromosoma segmento.
7.
Nessun delezioni o duplicazioni che coinvolgono GJB2 o GJB6 sono stati segnalati per causare perdita di udito non sindromica DFNA3 e sordità.
8.
Due varianti patogeniche in GJB6, p.Thr5Met e p.Ala40Val, sono stati riportati in una famiglia italiana e un individuo di Taiwan con DFNA3 [Grifa et al 1999, Yang et al 2007].
Geneticamente Correlate (alleliche) Disturbi
Altri fenotipi associati a mutazioni di GJB2 e GJB6 sono i seguenti:
GJB2
- DFNB1, un autosomica recessiva (o forse DIGENICA) condizione di (generalmente) moderato-grave compromissione neurosensoriale
- Cheratoderma palmoplantare con la sordità, caratterizzata da ipercheratosi diffusa delle mani e dei piedi[Richard et al 1998, Heathcote et al 2000, Feldmann et al 2005, de Zwart-Storm et al 2008]. Questa condizione è causata da una variante patogena eterozigote in GJB2.
- Cheratite-ittiosi-sordità (KID) sindrome, una displasia ectodermica caratterizzata da vascolarizzante cheratite, progressivo erythrokeratoderma e profonda perdita dell’udito neurosensoriale, così come alopecia cicatriziale e predisposizione al carcinoma a cellule squamose [Richard et al 2002, van Geel et al 2002, van Steensel et al2002]. La sindrome KID è causata da una variante patogena eterozigote in GJB2.
- Ittiosi-sordità (HID) Sindrome hystrix-like, un autosomica dominante ereditaria cheratinizzanti malattia caratterizzata da ipoacusia neurosensoriale e ipercheratosi della pelle. Poco dopo la nascita, eritrodermia sviluppa, con ipercheratosi appuntita e ciottoli, come di tutta la superficie della pelle che appaiono dall’età di un anno.Grave cheratoderma palmo-plantare e alopecia cicatriziale si verificano in alcuni. Sindrome HID è considerato diverso dalla sindrome KID in: (1) la misura e il tempo di comparsa di sintomi cutanei; (2) la gravità della cheratite; e (3) di elettroni caratteristiche microscopiche. Sindrome KID e la sindrome HID sono causati dalla stessa variante patogena in GJB2 [van Geel et al 2002].
- La sindrome Vohwinkel, un autosomica dominante condizione classificata come “mutilazione” cheratoderma diffusa a causa ipercheratosi circonferenziale delle cifre può portare a autoamputazione (definito “pseudoainhum”). Lieve a moderata ipoacusia neurosensoriale è spesso associata con la malattia. Sindrome Vohwinkel è causata da una variante patogena eterozigotica nel GJB2 [Maestrini et al 1999].
- Sindrome di Bart-Pumphrey (BPS), un autosomica dominante disturbo caratterizzato da cheratoderma palmo-plantare, pad nocca, leuconichia, e sordità neurosensoriale. Le caratteristiche cliniche in parte si sovrappongono con la sindrome di Vohwinkel e la sindrome KID. BPS è causata da una variante patogena eterozigote in GJB2[Richard et al 2004]. Nota: La p.Met34Thr variante patogena in GJB2 descritta in una famiglia con cheratoderma palmo-plantare e autosomica dominante sordità neurosensoriale [Kelsell et al 1997] non è una causa di perdita dell’udito dominante [Cucci et al 2000]. Questo stesso DNA variante è stata individuata in persone udito normale[Denoyelle et al 1998, Kelley ed altri 1998, Feldmann et al 2004], e uno schermo di 128 nonni o capi di singole famiglie non noti per essere correlati e inclusi nel CEPH (Centro d’Etude du Polymorphisme Humain) ha identificato tre persone (2,3%) con la variante patogena [dati non pubblicati].
Con le varianti patogeniche di GJB2 in cui la malattia epidermica e perdita di udito cosegregate, la gravità della malattia della pelle fenotipo è altamente variabile, suggerendo che altri fattori modificano gene espressione [Kelsell et al 2001,Feldmann et al 2005]. E ‘importante valutare clinicamente ogni paziente con autosomica dominante perdita di udito per tutte le caratteristiche sindromiche che possono essere stati trascurati in affetti familiari.
GJB6
- La sindrome Clouston, un autosomica dominante condizione caratterizzata da displasia ectodermica idrotica, alopecia, e palmoplantare ipercheratosi [Smith et al 2002]
Caratteristiche cliniche
Descrizione clinica
Perdita dell’udito non sindromica e sordità, DFNA3, è caratterizzata da progressiva, ad alta frequenza insufficienza uditiva neurosensoriale da lieve a grave. Prelinguale e insorgenza postlinguale di perdita di udito è riportata con varianti patogeniche DFNA3 associati a GJB2 mentre DFNA3 associata varianti patogeniche in GJB6 causare la perdita dell’udito prelinguale [Weegerink 2013]. Quando la perdita dell’udito è postlinguale, i pazienti con perdita dell’udito DFNA3-associati possono passare alla schermata uditivo neonatale. Audioprofiles DFNA3 possono variare in modo significativo, anche all’interno di una famiglia. Gli individui con DFNA3 hanno altre constatazioni mediche associate.
Prove di funzione vestibolare e la tomografia computerizzata delle ossa temporali nelle persone con DFNA3 sono normali [Denoyelle et al 2002].
GJB2. Le varianti patogeniche dodici missense e p.Cys202Phe) che causano la sordità al DFNA3 locus sono associati con almeno due audioprofiles diverse in base all’età di insorgenza.
La maggior parte delle varianti patogeniche causare la perdita dell’udito prelinguale (p.Trp44Cys, p.Pro58Ala, p.Arg75Gln, p.Arg75Trp, p.Arg143Gln e p.Arg184Gln):
- Il audioprofile p.Trp44Cys è caratterizzato da una perdita neurosensoriale bilaterale simmetrica che varia da lieve a profonda, cominciando con perdita dell’udito ad alta frequenza e progressione di perdita a tutte le frequenze.
- La perdita dell’udito legate alla variante patogena p.Pro58Ala è progressiva, che vanno da lievi a gravi.
- La perdita di udito associata alle p.Arg75Gln e p.Arg75Trp varianti patogeniche di solito è profonda (soglia media per p.Arg75Gln è di 105 dBHL).
- Gli individui con la variante patogena p.Arg143Gln mostrano una profonda perdita dell’udito ad alta frequenza progressiva.
- Audioprofiles per gli individui con il c.551G> A (p.Arg184Gln) variante patogena sono downsloping e coerenti con grave a profonda perdita dell’udito prelinguale [Janecke et al 2001, Löffler et al 2001, Tekin et al 2001,Denoyelle et al 2002 , Feldmann et al 2005, Primignani et al 2007, Weegerink et al 2011].
Al contrario, la sordità legata alle varianti patogeniche conseguente sostituzioni p.Thr55Asn, p.Asp179Asn, e p.Cys202Phe è postlinguale:
- Audioprofiles di individui con la c.164C> A (p.Thr55Asn) variante patogena hanno un modello downsloping e sono coerenti con una perdita uditiva grave postlinguale-to-profonda.
- Età di esordio per la perdita dell’udito negli individui con la variante p.Asp179Asn gamme patogeni dalla prima alla terza decade. Il audioprofile mostra una perdita uditiva lieve-moderata, soprattutto alle alte frequenze.
- La perdita dell’udito in individui con la variante patogena p.Cys202Phe di solito non è rilevata fino alla seconda decade. Inizialmente, la perdita colpisce di preferenza le alte frequenze, ma progredisce di influenzare le frequenze medie di mezza età [Morle et al 2000, Denoyelle et al 2002, Primignani et al 2003, Melchionda et al2005].
Altro:
- Audioprofiles per gli individui con il p.Trp44Ser sostituzione patogeno non sono disponibili.
- La sostituzione p.Asp46Asn mostra intrafamilial variabilità nei audioprofiles con alcuni individui che mostrano postlinguale perdita progressiva dell’udito con esordio nella prima decade di vita e un po ‘mostrando perdita dell’udito prelinguale.
- Il p.Met163Leu variante patogena causa una perdita dell’udito ad alta frequenza da lieve a moderata; età di esordio non è stato segnalato [Hamelmann et al 2001, Marziano et al 2003, Matos et al 2008, Bazazzadegan et al2011].
GJB6. Il p.Thr5Met variante patogena in GJB6 è stato riportato in una famiglia [Grifa et al 1999]. La audioprofile di questa famiglia è caratterizzata da middle di perdita dell’udito ad alta frequenza. Il grado di perdita dell’udito è progressiva e variabile, che vanno da lievi a profonde. Non è stata riportata l’età di insorgenza di perdita dell’udito.
Il p.Ala40Val sostituzione nel GJB6 è stato identificato in un individuo con autosomica dominante perdita di udito non sindromica; il audioprofile e l’età di insorgenza non sono stati riportati [Yang et al 2007, Wang et al 2011].
Genotipo-fenotipo Correlazioni
Vedere descrizione clinica.
Penetranza
Causative mutazioni nel GJB2 e GJB6 sono completamente penetranti. Gli individui che ereditano una dominantemutazione in GJB2 o GJB6 sviluppano perdita di udito non sindromica DFNA3 associata. Mutazioni recessive inGJB2 o GJB6 causa perdita DFNB1-associato di udito non sindromica.
Nomenclatura
La diversa gene loci per la sordità non sindromica sono designati DFN (per la sordità).
Loci sono denominati in base a modalità di trasmissione:
- DFNA: autosomica dominante
- DFNB: autosomica recessiva
- DFN: X-linked
Il numero che segue le designazioni di cui sopra riflette l’ordine di gene mappatura e / o la scoperta.
Prevalenza
La prevalenza relativa di DFNA3 come causa di autosomica dominante perdita di udito non sindromica non è nota, ma è estremamente raro. Quattordici varianti patogeniche sono state descritte in tutto il mondo. La maggior parte di queste varianti patogeniche sono descritti solo in singole famiglie o casi simplex (cioè, una singola occorrenza in una famiglia)[Denoyelle et al 2002, Hilgert et al 2009].
Prevalenza per le diverse varianti patogeniche varia in base alla popolazione [Abe et al 2000, Hamelmann et al 2001,Löffler et al 2001, Liu ed altri 2002, Xiao & Xie 2004].
Diagnosi differenziale
Altre cause di postlinguale, devono essere considerati (vedi forme acquisite di ipoacusia Sordità ed Ereditaria Hearing Loss Panoramica).
Autosomiche dominanti forme sindromiche di ipoacusia con:
- Malformazioni della testa e del collo. Branchiootorenal (BOR) La sindrome è caratterizzata da malformazioni del esterno, medio e orecchio interno associato con conduttiva, neurosensoriale, o compromissione dell’udito mista; fistole e cisti branchiale; e malformazioni renali che vanno da ipoplasia renale da lieve a agenesia renale bilaterale [Chang et al 2004]. Varianti patogeniche in EYA1 sono causali.
- Anomalie della pigmentazione. Waardenburg sindrome tipo 1 (WS1) è caratterizzata da congenita perdita neurosensoriale dell’udito e disturbi della pigmentazione dell’iride, capelli e pelle, insieme a distopia canthorum (spostamento laterale del angoli palpebrali interno) [DeStefano et al 1998]. La perdita dell’udito si verifica in circa il 57% ed è congenita, neurosensoriale, tipicamente non progressiva, e sia unilaterale o bilaterale. Più comunemente, la perdita dell’udito è bilaterale e profonda (> 100 dB). La maggior parte degli individui con WS1 hanno o un ciuffo bianco (45%) o ingrigimento dei capelli del cuoio capelluto prima dei 30 anni. Gli individui affetti possono avere completo iridio eterocromia, parziale / segmentale eterocromia, o ipoplasia o brillanti iridi azzurre. La diagnosi è stabilita dai dati clinici. I criteri diagnostici si basano sulla presenza di perdita dell’udito neurosensoriale, alterazioni della pigmentazione, e il calcolo dell’indice di W di identificare distopia canthorum.Varianti patogeniche in PAX3 sono causali.
Vedere Sordità, autosomica dominante: OMIM fenotipica Series, una tabella di fenotipi simili che sono geneticamente diverse.
Gestione
Le valutazioni dopo la diagnosi iniziale
Consigliato dopo la diagnosi di sordità non sindromica e sordità, DFNA3:
- Valutazione completa di acuità uditiva utilizzando test appropriati all’età, tra cui test ABR, uditivo risposta a regime di prova (ASSR), e / o audiometria tonale
- Consultazione Genetica medica
Trattamento delle manifestazioni
Il trattamento è indicato seguente:
- Raccordo con apparecchi acustici appropriati
- L’iscrizione in un programma educativo appropriato per i non udenti
- Esame di impianto cocleare, una opzione di abilitazione promettente per le persone con sordità profonda [Connellet al 2007]
- Il riconoscimento che, a differenza di molte condizioni cliniche, la gestione e il trattamento della sordità lieve a profonda rientrano in gran parte di competenza del benessere sociale e di sistemi d’istruzione, piuttosto che il sistema di assistenza medica [Smith et al 2005]
Prevenzione delle complicanze secondarie
La diagnosi precoce, abilitazione con apparecchi acustici o l’impianto cocleare, e la programmazione educativa diminuirà la probabilità di discorso a lungo termine o di ritardo scolastico.
Sorveglianza
I seguenti sono adatti:
- Esame semestrale da un medico che abbia familiarità con deficit uditivo ereditario
- Ripetere audiometria per confermare la stabilità di perdita dell’udito
Agenti / Circostanze da evitare
Gli individui con perdita dell’udito dovrebbero evitare esposizioni ambientali noti per causare la perdita dell’udito. Più importante è evitare l’esposizione ripetuta a rumori forti.
Valutazione del Parenti a rischio
Test di genetica molecolare è consigliato per a rischio parenti di individui con una nota variante patogena. Gli individui con note varianti patogeniche dovrebbero essere seguite semestralmente da un medico che abbia familiarità con deficit uditivo ereditario.
Raccomandazioni per la valutazione dei membri a rischio della famiglia quando i test genetici molecolari non è disponibile includono audiometria tonale per valutare l’acuità uditiva e la revisione della storia clinica e l’esame fisico per escludere altri risultati sistemici.
Vedere consulenza genetica per le questioni legate alla sperimentazione di a rischio parenti per consulenza geneticascopi.
Terapie Sotto inchiesta
Cerca ClinicalTrials.gov per l’accesso alle informazioni su studi clinici per una vasta gamma di malattie e condizioni.Nota: Non ci può essere la sperimentazione umana per questa condizione.
Consulenza genetica
La consulenza genetica è il processo di fornitura di individui e famiglie con informazioni sulla natura, l’ereditarietà, e le implicazioni di malattie genetiche per aiutarli a prendere decisioni personali e mediche informate. I seguenti sezione tratta la valutazione del rischio genetico e l’uso della storia familiare e test genetici per chiarire lo status genetico per i familiari. Questa sezione non è destinata a affrontare tutte le questioni personali, culturali, etiche o che gli individui possono affrontare o per sostituire la consultazione con una genetica professionali. -ED.
Modalità di eredità
Perdita di udito non sindromica e sordità, DFNA3, è ereditata come autosomica dominante maniera.
Rischio per i familiari
I genitori di un probando
- La maggior parte delle persone con diagnosi di DFNA3 avere un genitore sordo; la storia della famiglia è raramente negativo.
- Un probandi con DFNA3 può avere la condizione come il risultato di de novo mutazioni di GJB2 o GJB6. La proporzione di casi causati da de novo mutazione è molto piccola.
- Raccomandazioni per la valutazione dei genitori di un probando con un apparente de novo variante patogena comprendono (a) la valutazione di acuità uditiva utilizzando test ABR emissioni e audiometria tonale e (b) l’anamnesi e l’esame fisico per determinare se sono presenti altri risultati sistemici.
Fratelli e sorelle di un probando
- Il rischio di fratelli e sorelle dipende dallo stato genetico dei probando genitori ‘s.
- Se uno dei probando genitori ‘s ha un GJB2 o GJB6 variante patogena, ogni sib ha una probabilità del 50% di ereditare la mutante allele.
- Quando i genitori sono clinicamente inalterati, il rischio per i fratelli e sorelle di un probando sembra essere basso.
- Se una variante patogena non può essere rilevato nel DNA estratto da leucociti di entrambi i genitori, due possibili spiegazioni sono mosaicismo germinale in un genitore o de novo mutazione nel probando. Sebbene non siano stati riportati casi di mosaicismo germinale, rimane una possibilità.
Figli di un probando. Figli di un affetto individuo hanno una probabilità del 50% di ereditare la GJB2 o GJB6variante patogena.
Altri membri della famiglia di un probando
- Il rischio per gli altri membri della famiglia dipende dallo stato delle probando genitori ‘s.
- Se un genitore è sorda, il suo membri della famiglia sono a rischio.
Genetica Related Issues Counseling
Vedere Gestione, Valutazione di parenti a rischio per le informazioni sulla valutazione a rischio parenti ai fini della diagnosi precoce e del trattamento.
Stabilire durante l’infanzia o nella prima infanzia se un bambino a rischio ha ereditato la alterato GJB2 o GJB6 alleledeve essere considerato in modo che il supporto e la gestione adeguata e precoce possono essere forniti per il bambino e la famiglia. Test di genetica molecolare per un GJB2 o GJB6 variante patogena può essere considerata solo se una variante patogena in entrambi gene è stato identificato in un colpiti membro della famiglia.
Ulteriori punti da considerare sono i seguenti:
- La comunicazione con le persone che sono sordi richiede i servizi di un interprete qualificato.
- Le persone sorde possono visualizzare la sordità come una caratteristica distintiva e non come un handicap, perdita di valore, o patologie che richiedono una “cura” o “cura”, o da “impedito”. Infatti, avere un bambino sordo può essere preferito avere un figlio con udito normale.
- Molte persone sorde sono interessati ad ottenere informazioni sulla causa della loro sordità informazioni anche per quanto riguarda i servizi sanitari, educativi e sociali piuttosto che informazioni circa la prevenzione, la riproduzione o la pianificazione familiare. Come in tutta la consulenza genetica, è importante per il consulente di individuare, riconoscere e rispettare / della famiglia dell’individuo domande, dubbi e paure.
- L’uso di alcuni termini è preferito: probabilità o possibilità vs rischio; non udenti e di udito vs udenti. Termini come “affetto”, “anormale” e “malattia che causa” dovrebbero essere evitati.
Pianificazione famigliare
- Il momento ottimale per la determinazione del rischio genetico e discussione della disponibilità di test prenatale è prima della gravidanza.
- È opportuno offrire una consulenza genetica (compresa la discussione dei rischi potenziali per la prole e le opzioni di riproduzione) di giovani adulti non udenti.
Considerazioni in famiglie con un apparente de novo variante patogena. Quando nessuno dei genitori di unprobando con una autosomica dominante condizione ha la variante patogena o evidenza clinica della condizione, ilGJB2 o GJB6 variante patogena è probabile de novo. Tuttavia, possibili spiegazioni non medici, tra cui la paternità alternativo o di maternità (ad esempio, con la riproduzione assistita) o l’adozione riservate potrebbero anche essere esplorate.
Banking DNA è la conservazione di DNA (tipicamente estratto da globuli bianchi) per un possibile uso futuro. Poiché è probabile che la metodologia di prova e la nostra comprensione dei geni, varianti alleliche, e le malattie migliorerà in futuro, occorre tenere in considerazione per bancaria DNA di affetti individui.
Test prenatale
Se il GJB2 o GJB6 variante patogena è stata identificata in un colpiti membro della famiglia, test prenatale per gravidanze a rischio può essere disponibile da un laboratorio clinico che offre sia il test del gene di interesse o test prenatale personalizzato.
Le richieste di test prenatale per condizioni come DFNA3 non sono comuni. Possono esistere differenze di prospettiva tra i professionisti medici e all’interno delle famiglie per quanto riguarda l’uso di test prenatali, in particolare se il test viene presa in considerazione ai fini della interruzione della gravidanza, piuttosto che la diagnosi precoce. Sebbene la maggior parte centri prenderebbero in considerazione decisioni su test prenatale per essere la scelta dei genitori, la discussione di questi problemi è appropriato.
Diagnosi genetica preimpianto (PGD) può essere un’opzione per alcune famiglie in cui il GJB2 o GJB6 è stato identificato variante patogena.
Genetica molecolare
Le informazioni contenute in tabelle Genetica Molecolare e OMIM può differire da quello in altre parti del GeneReview: tavoli può contenere informazioni più recenti. – ED.
Tabella A.
Non sindromica, udito e Sordità, DFNA3: Geni e database
|
Locus Nome |
Gene |
Cromosoma Locus |
Proteina |
Locus specifico |
HGMD |
|
DFNA3 |
La homepage Connessina-sordità (GJB2) |
||||
|
DFNA3 |
La homepage Connessina-sordità (GJB6) |
I dati sono compilati dai seguenti riferimenti standard: gene da HGNC; cromosoma locus, nome luogo, regione critica, complementazione gruppo da OMIM; proteine da UniProt. Per una descrizione di database (Locus specifico, HGMD) a cui vengono forniti i link, clicca qui.
Tabella B.
OMIM Voci per non sindromica, udito e sordità, DFNA3 (Visualizza tutto in OMIM)
|
GAP JUNCTION PROTEINA, BETA-2; GJB2 |
|
|
SORDITA ‘, AUTOSOMICA 3A DOMINANTE; DFNA3A |
|
|
GAP JUNCTION PROTEINA, BETA-6; GJB6 |
|
|
SORDITA ‘, autosomica dominante 3B; DFNA3B |
GJB2
Struttura genica. La maggior parte connexine hanno un’architettura comune, con l’intera regione codificante contenuta in un unico grande esone separato dalla regione 5′-non tradotta da un introne di dimensioni variabili. La sequenza codifica di GJB2 (esone 2) 681 paia di basi (compreso l’arresto codone) e si traduce in una proteina di 226-amino, connessina 26 (Cx26). Per un riepilogo dettagliato delle gene informazioni e proteine, vedi tabella A, Gene.
Varianti alleliche benigni. Numerosi alleli benigni GJB2 sono stati segnalati e sono quotate sul Connessina-Sorditàpagina.
Varianti alleliche patogeni. Ci sono 12 note varianti patogeniche in GJB2 (vedi tabella 2). La maggior parte di queste varianti hanno dimostrato di separare in famiglie; tuttavia, i p.Arg75Gln e p.Arg75Trp varianti patogeniche di GJB2sono anche stati identificati come de novo varianti in casi simplex (cioè, una singola occorrenza in una famiglia). Queste due varianti patogeniche sono implicati sia autosomica dominante perdita di udito non sindromica e perdita di udito sindromica associata a disturbi della pelle (vedi disturbi geneticamente correlate [Janecke et al 2001, Feldmann et al2005].
La patogenicità del p.Arg75Trp variante è stata messa in discussione, come è stato riportato in uno dei 77 controlli egiziani il cui status udienza non è stata riportata [Richard et al 1998]. Tuttavia, i rapporti successivi casi, modelli animali e studi funzionali sostengono con forza per la patogenicità di questa variante [Janecke et al 2001, Kudo et al2003, Maeda et al 2005, Maeda et al 2009, Mani et al 2009, Yum et al 2010 , Weegerink et al 2011, Zhang et al 2011].
Tabella 2.
Selezionata GJB2 allelica Varianti
|
Variante Classificazione |
DNA Nucleotide Change |
Proteine Amino Acid Change |
Sequenze di riferimento |
|
Benigno |
c.101T> C |
p.Met34Thr 1 |
|
|
Pathogenic |
c.132G> C |
p.Trp44Cys |
|
|
c.136G> A |
p.Asp46Asn |
||
|
c.164C> A |
p.Thr55Asn |
||
|
c.131G> C |
p.Trp44Ser |
||
|
c.172C> G |
p.Pro58Ala |
||
|
c.223C> T |
p.Arg75Trp |
||
|
c.224G> A |
p.Arg75Gln |
||
|
c.428G> A |
p.Arg143Gln |
||
|
c.487A> C |
p.Met163Leu |
||
|
c.535G> A |
p.Asp179Asn |
||
|
c.551G> A |
p.Arg184Gln |
||
|
c.605G> T |
p.Cys202Phe |
Nota sulla classificazione variante: Varianti elencate nella tabella sono stati forniti dagli autori. GeneReviews personale non ha verificato in modo indipendente la classificazione delle varianti.
Nota sulla nomenclatura: GeneReviews segue le convenzioni di denominazione standard del Human Genome Variation Society (www .hgvs.org). Vedere di riferimento rapido per una spiegazione di nomenclatura.
1.
Variante trovato nelle persone udito normale e familiari con cheratoderma palmo-plantare
Normale gene prodotto. Connexin 26 è una proteina beta-2 gap junction. Giunzioni sono organelli altamente specializzati, comprensivi di canali cluster che permettono lo scambio intercellulare diretto di ioni e molecole attraverso i pori acquosi centrali. Ruoli postulate includono la rapida propagazione di segnali elettrici e la sincronizzazione di attività in tessuti eccitabili e lo scambio di metaboliti e molecole segnale nei tessuti non eccitabili [Evans & Martin2002].
Ogni proteina connessina contiene due extracellulare (E1-E2), quattro transmembrana (M1-M4), e tre domini citoplasmatici (N-terminale, C-terminale, e un ciclo citoplasmatica situato tra M2 e M3) [Maeda et al 2009]. Ogni dominio extracellulare contiene tre residui di cisteina uniti tra E1 ed E2 loop da almeno un legame disolfuro [Kovacs et al 2007, Yeager & Harris 2007]. La presunta importanza di questi sei cisteine si può dedurre da esperimenti CX32 in cui ogni Cys variante patogena blocca completamente lo sviluppo di conduttanze gap-giunzione tra Xenopus coppie ovociti. Il dominio transmembrana terzo (M3) è anfipatico e riveste la parete putativo del canale intercellulare [Kovacset al 2007, Yeager e Harris 2007], che è stato creato per oligomerizzazione di sei connessine a formare una struttura esamerica chiamata connessone. Due connexons, uno da ciascuna cella, si uniscono nel tagliato extracellulare per completare il percorso cellula-cellula. Se i connexons fornite da ciascuna cella sono di identica composizione, il canale è omotipica; se ogni connessone è formato da una diversa composizione di connessine, è definito eterotipica. La maggior parte delle connessine sono fosfoproteine e subiscono modificazioni post-trascrizionali [Moreno 2005, Locke et al2006]. Cx26 forma combinazioni funzionali con sé, Cx30, Cx31, Cx32, Cx46, e CX50 [Cottrell e Burt 2005, Liu ed altri 2009].
Anormale gene prodotto. Canali Gap di giunzione sono permeabili a ioni e piccole metaboliti con masse molecolari relative fino a circa 1,2 kd [Harris & Bevans 2001]. Le differenze nei meccanismi di selettività e di gating ionici tra giunzioni riflettono l’esistenza di oltre 20 diversi connessina isoforme negli esseri umani.
L’anormale prodotto del gene in DFNA3 provoca sordità attraverso un dominante meccanismo -negativa d’azione. La maggior parte delle varianti patogeniche in GJB2 sono stati funzionalmente testata per gli effetti dominanti negativi in sistemi di espressione ricombinanti (p.Trp44Cys, p.Trp44Ser, p.Arg75Trp, p.Arg75Gln, p.Arg143Gln, p.Met163Leu, p.Asp179Asn, p.Arg184Gln e p.Cys202Phe). La capacità di prevenire la formazione di canali di giunzione gap funzionale è stata dimostrata in primo luogo con la p.Arg75Trp variante patogena in un Xenopus sistema modello dell’ovocita [Richard et al 1998, Mani et al 2009]. I p.Trp44Cys, p.Trp44Ser, e p.Arg75Gln varianti patogeniche hanno dimostrato di prevenire la formazione del canale funzionale in vitro [Bruzzone et al 2001, Marziano et al 2003, Piazza et al 2005]. Oltre alla inibizione dominante negativo di wild-type Cx26, la maggior parte delle varianti patogeniche (p.Trp44Cys, p.Trp44Ser, p.Arg75Trp, p.Arg75Gln, p.Arg143Gln, p.Asp179Asn, p.Arg184Gln, e p. Cys202Phe) mostrano anche un effetto trans-dominante negativo on wild-type formazione del canale Cx30. Il p.Met163Leu variante patogena mostra un effetto dominante negativo adeguati al traffico di proteine e la vitalità delle cellule [Matos et al2008]. I display variante Thr55Asn compromessa la tratta e non riesce a raggiungere la membrana plasmatica[Melchionda et al 2005].
GJB6
. Struttura Gene La maggior parte dei geni giunzione gap ha due esoni; pochi hanno un solo esone, e uno, GJB6, ha tre esoni, di cui solo la terza è di codifica. La proteina tradotto, connessina 30 (Cx30), è lunga 261 aminoacidi. Per un riepilogo dettagliato delle gene informazioni e proteine, vedi tabella A, Gene.
Varianti alleliche patogeni. Ci sono solo due noti DFNA3 causando varianti patogeniche di GBJ6: p.Thr5Met e p.Ala40Val [Grifa et al 1999, Yang ed altri 2007] (vedi tabella 3).
Tabella 3.
Selezionata GJB6 patogeno allelica Varianti
|
DNA Nucleotide Change |
Proteine Amino Acid Change |
Sequenze di riferimento |
|
c.14C> T |
p.Thr5Met 1 |
|
|
c.119C> T |
p.Ala40Val |
Nota sulla classificazione variante: Varianti elencate nella tabella sono stati forniti dagli autori. GeneReviews personale non ha verificato in modo indipendente la classificazione delle varianti.
Nota sulla nomenclatura: GeneReviews segue le convenzioni di denominazione standard del Human Genome Variation Society (www .hgvs.org). Vedere di riferimento rapido per una spiegazione di nomenclatura.
1.
Vedi anche Tabella 1 e descrizione clinica, GJB6.
Normale gene prodotto. Connexin 30 è una proteina beta-6 gap junction. Condivide un’architettura che è comune a tutte le connessine (vedi GJB2, prodotto genico Normale).
Anormale prodotto del gene. Come i prodotti genici anormali di GJB2 in DFNA3, le varianti p.Thr5Met e p.Ala40Val di GJB6 atto attraverso un dominante meccanismo -negativa di inibire l’attività di wild-type canali gap Cx30 giunzione. Inoltre, la variante p.Ala40Val esercita un effetto trans-dominante negativo sulla Cx26, alterando gap junction formazione nella coclea. Negli studi funzionali con un modello di topo, la variante p.Thr5Met ha dimostrato di esercitare il suo effetto patogeno attraverso accoppiamento biochimico diminuito tra le cellule cocleari [Grifa et al 1999,Schütz et al 2010, Wang et al 2011].
La perdita dell’udito e connessina 26
Martijn H Kemperman, MD, Bugie H Hoefsloot, PhD, e 1 Cor WRJ Cremers, MD PhD
Informazioni Autore ►copyright e licenza ►
Questo articolo è stato citato da altri articoli in PMC.
Ipoacusia è una disabilità sensoriale che colpisce milioni di persone in tutto il mondo. Anche se in pericolo di vita, non si può diventare un peso importante nella vita sociale e professionale. Nel mondo industrializzato, sordità di origine infettiva e / o ambientale è diventato meno frequenti, con un conseguente aumento della percentuale di deficit uditivo ereditario. Sordità si verifica in 1: 1000 neonati 1 e la causa è ereditaria in circa la metà. Questo tipo di ipoacusia è a volte indicato come prelinguale, quanto influisce il bambino prima dell’età di sviluppo del linguaggio. Una distinzione può essere fatta tra sordità sindromica, in cui la sordità è accompagnata da altre anomalie specifiche, e sordità non sindromica (circa 75%), in cui non ci sono ulteriori anomalie. Circa tre quarti delle forme non sindromiche sono causati da una malattia recessiva 1,2,3,4.La tabella 1 fornisce una panoramica di alcune caratteristiche epidemiologiche.
Caratteristiche epidemiologiche di perdita dell’udito prelinguale
Tra il 1997 e oggi, molte forme ereditarie non sindromiche di sordità sono stati localizzati sul genoma umano mediante tecniche di linkage genetico. A seconda del modello di ereditarietà della sordità, questi loci sono designati DFNA (autosomica dominante), DFNB (autosomica recessiva) o DFN (X-linked). Essi sono numerati in ordine cronologico di scoperta. Per la maggior parte di questi loci non sono stati identificati i geni che causano malattie sottostanti finora. In udienza ereditario Loss Homepage 5 sono riassunte tutte queste forme attualmente conosciuti di sordità ereditaria. Le tabelle Tables22,33,44,5,5, derivato da questo homepage, illustrano i risultati ottenuti in questo campo di ricerca. Alcuni gruppi di ricercatori, dopo aver trovato prove preliminari di un nuovo locus, hanno sostenuto (‘riservato’) loci in anticipo. ‘Ritirato’ indica quelli che si è rivelato non essere corretto. La maggior parte di questi tipi genetici di ipoacusia sono abbastanza rari, con l’eccezione di DFNB1. Questo documento affronta DFNB1, che è causata da mutazioni nel gene connessina 26.
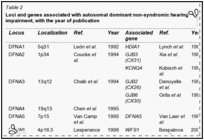
Loci e geni associati con autosomica deficit uditivo non sindromica dominante, con l’anno di pubblicazione
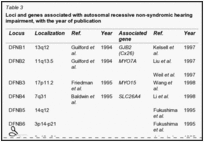
Loci e geni associati con autosomica deficit uditivo non sindromica recessiva, con l’anno di pubblicazione
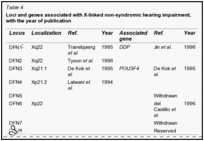
Loci e geni associati con deficit uditivo non sindromica X-linked, con l’anno di pubblicazione
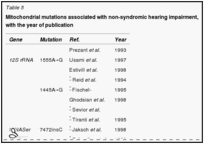
Mutazioni mitocondriali associate a deficit uditivo non sindromica, con l’anno di pubblicazione
PROBLEMA DI UDITO
Sebbene il gene GJB2 connessina 26 è anche coinvolta in una forma autosomica dominante di sordità (DFNA3), la maggior parte delle mutazioni in questo gene causa recessiva sordità / ipoacusia bilaterale ereditaria, cosiddetti DFNB1. Questa forma di perdita di udito non sindromica neurosensoriale è prelinguale e la sua gravità varia da lieve a profonda, in una certa misura a seconda del tipo di mutazione 6,7. La perdita dell’udito nella fascia alta tono è stato recentemente descritto come un aspetto caratteristico, ma tutte le frequenze sono interessate 8. In due terzi dei casi, la perdita di udito non è progressiva e non ci sono di solito non vestibolare e / o anomalie labirintiche.
GENETICA
DFNB1 è stato il primo locus incriminato in autosomica recessiva sordità; nel 1997 GJB2 è risultato essere responsabile 9 GJB2 è un piccolo gene situato sul cromosoma 13q11.; ha una lunghezza di circa 5,5 kilobasi.Ci sono due esoni, di cui solo uno contiene la sequenza codificante. L’mRNA è lungo 2,4 chilobasi e si traduce in una proteina con 226 aminoacidi. Questa proteina appartiene alla famiglia connessine, che attualmente ha più di una dozzina di membri 10.
LA PROTEINA (figura 1)
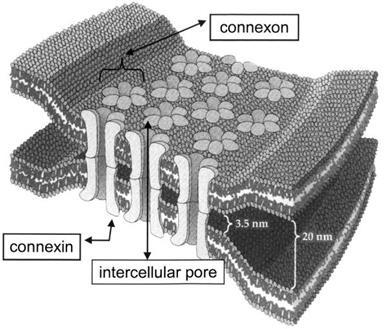
Rappresentazione schematica di una giunzione gap. Sei connessine formano una connessone. Due connexons di celle vicine formano i pori, che permettono il trasporto intercellulare di piccole molecole (Adattato da Rif. 22)
Le connessine sono proteine di membrana con quattro domini transmembrana. Sei catene di queste proteine formano un complesso (un esamero), chiamato connessone. Due esameri nelle membrane delle cellule adiacenti formano un canale cellula-cellula, una cosiddetta giunzione gap, che permette il trasporto di piccole molecole e ioni tra cellule. Un esamero può contenere vari tipi di connexin, e vari tipi di esamero possono formare canali cellula-cellula. I costituenti del canale determinano quali molecole o ioni possono passare attraverso 11.
LA CELLULA (FIGURA 2)
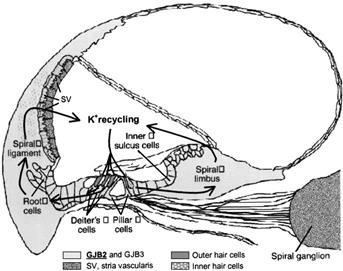
Sezione schematica attraverso la coclea umana che mostra K +riciclaggio percorso e l’espressione della connessina 26 (GJB2).(Adattato da Rif. 23)
Recentemente, l’ipotesi è stata avanzata che la proteina CX26 è essenziale per mantenere l’alta concentrazione di K + nel endolinfa dell’orecchio interno. Stimolazione sonora della catena degli ossicini causa vibrazioni nel endolinfa. Ioni K + entrano nelle cellule cigliate sotto l’influenza di queste vibrazioni e il segnale di vibrazione viene infine trasformato in un segnale neurale. Il sistema viene rigenerato dal rilascio di K + delle cellule ciliate nelle cellule di sostegno. Gli ioni K + sono poi passati da una cella all’altra attraverso giunzioni e sono poi rilasciati nel endolinfa. Eccetto per le cellule neurosensoriale, la proteina CX26 è presente in giunzioni comunicanti tutti i tipi cellulari presenti nella coclea, compreso il limbus a spirale, le cellule di supporto, il legamento a spirale e basale e cellule intermedie del vascularis stria. È quindi molto probabile che connessina 26 è coinvolto in K + -Riciclo nella coclea 11.
EPIDEMIOLOGIA
Mutazioni nel CX26 sono la causa più comune di sordità recessiva autosomica tutto il mondo. Questo gene è creduto rilevanti per la metà di tutti i casi di sordità ereditaria. CX26 mostra diverse mutazioni, ma una mutazione si verifica molto spesso in Europa, la mutazione 35delG. Frequenza media portante in Europa è 01:51 (nord / centro dell’Europa 1:79, sud Europa 1,35) 12(Tabella 6). Nei paesi mediterranei la frequenza portante supera persino quello della mutazione AF508 nel gene CFTR che causa la fibrosi cistica. Frequenze portanti in Nord America e in Australia sono paragonabili a quelli del Nord / Europa centrale. Nelle popolazioni orientali e gli ebrei Ashkenazi, altre mutazioni nello stesso gene svolgere un ruolo più importante (234delC 13 e 176delT 14, rispettivamente). L’alta frequenza di-connexin-26 relativa udienza di discussione compromissione in alcune popolazioni può essere il risultato della tradizione dei matrimoni tra persone udenti15. La mutazione 35delG dà luogo ad una gravemente ridotta, proteina non funzionale 16. Più di sessanta altro, molto meno frequente, le mutazioni sono state descritte in CX26 17. L’incertezza circa la patogenicità di alcuni delle mutazioni complica l’interpretazione di analisi di mutazione 18.

Frequenza portante di mutazioni nel gene 35delG GJB2 in 17 Paesi europei (adattato da Rif. 12)
Denoyelle et al. 7 hanno trovato mutazioni nel gene CX26 nel 49% delle famiglie provenienti da Francia, Gran Bretagna e Nuova Zelanda che hanno avuto gravi di profonda perdita dell’udito prelinguale. MutazioniCX26 erano presenti nel 51% del gruppo con, rispetto al 31% in il gruppo senza una chiara storia familiare di ipoacusia; 86% delle mutazioni CX26 erano mutazioni 35delG. Mueller et al. 19 ha studiato un gruppo di 284 pazienti inglesi con la prima infanzia deficit uditivo o sordità, con e senza cause ereditarie. Essi trovarono mutazioni CX26 nel 27,8% dei casi familiari e nel 7,9% dei casi sporadici; Il 70% delle mutazioniCX26 erano mutazioni 35delG. Questa differenza può essere spiegata con il fatto che le famiglie con diverse origini etniche sono stati inclusi nello studio. La prevalenza di non-familiare, disabilità uditiva sporadici sulla base di mutazioni CX26 in una popolazione inglese-belga di 68 bambini è stata del 10% 20.
DIAGNOSI
Un numero crescente di centri medici in grado di eseguire analisi di mutazione per determinare il coinvolgimento del gene CX26 in caso di compromissione dell’udito congenito. Questo metodo è stato disponibile per diversi anni presso il dipartimento di genetica medica a Nijmegen. Abbiamo analizzato retrospettivamente l’esito di novanta-uno CX26 richieste di analisi di mutazione per un periodo determinato di tempo. Diciannove casi indipendenti hanno mostrato di avere due mutazioni nel gene. Dodici di loro si è rivelato essere omozigote, mentre altri quattro erano eterozigoti per la mutazione 35delG. Nel complesso, la mutazione 35delG è stato coinvolto nel 84% dei casi; tredici casi provenivano da famiglie multiaffected, mentre altri tre sono stati casi sporadici. Le informazioni relative ai rimanenti tre famiglie non può essere recuperata. Tabella 7 fornisce una panoramica delle mutazioni CX26 si trovano a Nijmegen.
Panoramica delle mutazioni CX26 trovato a Nijmegen
Analisi mutazione non si applica solo ai bambini con una storia familiare chiaro, ma anche per i bambini i cui genitori hanno un udito normale (casi sporadici). Inoltre, se una mutazione in CX26 è presente, la consulenza genetica può essere offerto per fornire informazioni sulle risposte eziologia e il rischio di reiterazione in futuro prole. Quando una mutazione analisi è positiva ci sarà solitamente alcuna necessità di ulteriori indagini come l’imaging e test oftalmologici, perché altre cause di sordità congenita non devono più essere esclusi. In questi casi, l’attenzione può essere immediatamente focalizzata sull’ottimizzazione udito del bambino.L’esame istopatologico della coclea in un paziente con mutazione CX26 confermato ha rivelato un nervo acustico intatto 21. Ciò significa che questi pazienti sono candidati idonei per l’impianto cocleare, a condizione che la loro perdita di udito è sufficientemente profondo. La diagnosi precoce porta a un trattamento precoce, che dà i migliori risultati con l’impianto cocleare.
CONCLUSIONE
A differenza di molti altri geni CX26 è piccolo, in modo che lo screening per le mutazioni è veloce e relativamente semplice. Inoltre, l’elevato coinvolgimento complessivo di mutazioni CX26 in autosomiche recessive forme non sindromiche di sordità, e anche in casi sporadici, rende l’analisi di mutazione decisamente la pena-mentre. CX26 analisi di mutazione ha quindi assicurato un posto come uno strumento utile nella pratica clinica. Finora, molte diverse mutazioni nel gene CX26 causando DFNB1 sono stati identificati 17. L’incertezza circa la patogenicità della mutazione richiede una stretta collaborazione con i genetisti che hanno familiarità con la sordità 18. Tuttavia, l’analisi di mutazione CX26 offre un buon punto di partenza per la diagnosi molecolare dei pazienti con non sindromica sordità congenita.
Ringraziamenti
Questo lavoro è stato sostenuto dalla Organizzazione olandese per la ricerca scientifica, del Consiglio per la ricerca medica e sanitaria (progetto n 920-03-100) e la Fondazione ENT-Research Nijmegen, Paesi Bassi. Il testo si basa su un articolo pubblicato nel Nederlands Tijdschrift Voor geneeskunde.