DISORDINI VASCOLARI DELL’ORECCHIO INTERNO (non genetici)
GENERALITÀ
Per lungo tempo la sordità neurosensoriale è stata considerata la conseguenza dell’invecchiamento, dell’ototossicità, dell’esposizione cumulativa a rumore. Di fatto gli elementi cellulari della coclea tendono ad “accumulare” nel tempo le conseguenze di numerose “noxae” patogene. a cui vengono esposte le cellule gustative vivono per 14-21 giorni, le olfattive per 30-90 giorni. Le cellule cigliate dell’organo del Corti sono programmate nell’uomo per una vita di 70-100 anni, ma il loro numero, poco meno di 20000, è molto piccolo se confrontato con i 137 milioni di cellule della retina
Le strutture anatomiche all’interno della coclea risultano piuttosto inaccessibili ai mezzi di esplorazione solitamente utilizzati in patologia, quali ad esempio prelievo di liquidi o di tessuti Di conseguenza le conoscenze sui meccanismi patologici responsabili di numerose forme di cocleopatia sono ancora imprecise. Molto spesso la diagnosi di queste entità cliniche non è causale ma necessariamente solo descrittiva di alcuni aspetti audiometrici (grado si simmetria di soglia, presenza di recruitment, profilo di soglia, evoluzione temporale dell’ipoacusia ad es improvvisa, progressiva, fluttuante) e di sintomi concomitanti (vertigine ed acufeni) Sicuramente alcune forme di sordità cocleare, che condividono caratteristiche sintomatologiche ed audiometriche comuni sono in realtà determinate da agenti causali di diverso tipo il prototipo di queste forme può essere la sordità in corso di malattia di Ménière, la cui genesi è stata via via sostenuta invocando praticamente tutti i fattori patologici conosciuti, fatta forse eccezione per i fattori tumorali D’altra parte le conoscenze più recenti sulla fisiologia e sulla biochimica della coclea hanno evidenziato alcune peculiarità, che in qualche misura spiegano la difficoltà di associare alla sordità uno specifico fattore patologico. La salute delle strutture contenute nel dotto cocleare è basata su un delicato equilibrio omeostatico, principalmente regolato da processi locali. Le cellule cigliate rappresentano un grado di estrema specializzazione per un neuroepitelio, ed il loro metabolismo è regolato oltre che dal bagno dell’endolinfa, da uno scambio di informazioni biochimiche con le cellule di supporto A sua volta l’endolinfa è prodotta e regolata dalla stria vascolare, anch’essa una struttura altamente specializzata nel sostenere cicli metabolici molto veloci Sostanze tossiche possono entrare nella coclea attraverso la via ematica, causando alterazioni sia sull’apparato neuro-epiteliale responsabile della trasduzione meccano-elettrica, sia sulla struttura vascolare responsabile del mantenimento dell’omeostasi endolinfatica L’orecchio interno è anche capace di risposte immuno-mediate, in conseguenza della penetrazione di antigeni di origine virale o batterica; inoltre lo stesso orecchio interno è forse un organo bersaglio in alcune malattie auto-immuni.
Date queste assunzioni, i disordini che si sviluppano all’interno della coclea possono essere descritti, almeno per lo stato attuale delle conoscenze, come sostenuti da fattori di tipo genetico (vedi capitolo specifico), vascolare, flogistico-immunitario, dismetabolico. tossico.
FATTORI VASCOLARI
Parole chiave: interno orecchio, coclea, l’udito, vascolare
Le sordità per le quali più spesso si invoca una causa vascolare sono forme ad esordio improvviso (sordità improvvisa idiopatica), e forme lentamente progressive, spesso interpretate come presbiacusia. Nella sordità improvvisa il rapido esordio dei sintomi è riconducibile a fenomeni endovasali (microtrombi. impilamento del globuli rossi) o a lesioni delle pareti dei vasi (microemorragie. spasmi della muscolatura arteriolare). Qualsiasi fattore sistemico che coinvolge la circolazione generale può essere all’origine di questi fenomeni locali: malattia arteriosclerotica generalizzata, malattie ematologiche come leucemia, macroglobulinemia, policitemia, anemia falciforme. diabete, malattie autoimmuni. Anche una malattia infettiva come la sifilide, può causare una riduzione del flusso ematico locale dovuta a fenomeni di arterite obliterativa, concomitanti ad un alterazione del riassorbimento endolinfatico, Inoltre un meccanismo di vasospasmo è stato ipotizzato per spiegare alcuni casi di sordità improvvisa insorta in pazienti con cefalea.
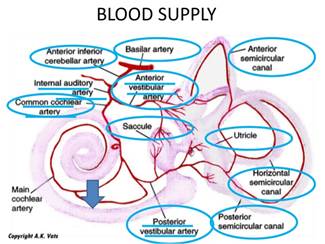
Le arterie interessate nei fenomeni cocleari ischemici sono l’arteria cerebellare antero-inferiore (AICA), la a. uditiva interna, l’a. cocleare principale con le branche vestibolo-cocleari, e la rete di piccole arterie spirali e radiali che si adattano al decorso della coclea (Fig.1 ),
|
|
|


Lo schema riporta la più comune variante della circolazione arteriosa che assicura l’irrorazione dell’orecchio interno, Dall’arteria basilare posta sulla faccia anteriore del tronco si diparte l’arteria cerebellare anteroinferiore (AICA) che origina l’arteria labirintica (anche detta arteria uditiva interna), Essa si suddivide in rami con scarse anastomosi che irrorano rispettivamente la coclea ed il labirinto posteriore. L’occlusione acuta dell’arteria labirintica è responsabile delle forme di ipoacusia pantonale improvvisa severa-profonda, accompagnate da vertigine, difficilmente reversibili. E’ probabile che in dipendenza del livello in cui avviene l’occlusione si possano determinare sordità di diversa entità e configurazione (es alte o basse frequenze), e associate o meno ad altri sintomi, espressione di danno ischemico nei distretti vicini (es, vertigine, paresi facciale). Il rallentamento di flusso e la riduzione di calibro della rete arteriosa cocleare causate da processi arteriosclerotici cronici sono responsabili di cocleopatie a lenta evoluzione.
L’anossia acuta della coclea è responsabile di variazioni del potenziale microfonico e del potenziale d’azione del nervo 8°, che avvengono in pochi minuti. La grande suscettibilità della coclea all’ipo-ossigenazione è determinata in parte dal suo elevato metabolismo ed in parte dalla circolazione cocleare che ha scarse possibilità di sviluppare circoli collaterali (circolazione “quasi-terminale”) Un ridotto apporto di ossigeno, se protratta nel tempo, causa un danno alle costituenti lipidoproteiche e al DNA delle cellule cigliate, cui segue la morte della cellula per necrosi (rottura della cellula) o per apoptosi (morte programmata). Protraendosi lo stato anossico il danno alle cellule cigliate diventa permanente, e quindi la sordità irreversibile, Un’evoluzione lentamente progressiva della vasculopatia è stata associata ad una atrofia variamente estesa della stria vascolare la quale sarebbe a sua volta responsabile di un insieme di processi degenerativi dell’organo del Corti. Questo aspetto, osservabile particolarmente durante l’età senile, definisce, secondo un criterio anatomo-patologico, una forma “vascolare” della presbiacusia.
Il recupero della funzione uditiva e vestibolare nelle forme di sordità improvvisa è in relazione alla possibilità o meno che venga ripristinata una sufficiente perfusione ematica del labirinto cocleo-vestibolare,
PROBLEMI CIRCOLATORI Qualsiasi interferenza con la circolazione delle delicate strutture dell’orecchio interno o delle loro connessioni centrali può provocare una vertigine, e talvolta anche sordità ed acufeni (sensazione soggettiva di rumore nell’orecchio). Queste modificazioni della circolazione possono essere il risultato di uno spasmo dei vasi, di una loro parziale o totale occlusione, o rottura con emorragia.
· a) Spasmo: La vertigine periferica (dell’orecchio interno) causata da uno spasmo dei vasi sanguigni, si presenta in genere all’improvviso, ed è di tipo intermittente. Le cause che possono dare tale vertigine sono la fatica intellettuale e gli stress emotivi. Alcune droghe come ad esempio la caffeina (caffè e thè) e la nicotina (sigarette) possono provocare uno spasmo vascolare e dovrebbero quindi essere ridotte ed evitate.
· b) Stenosi: Con l’età i vasi sanguigni diventano più spessi e più duri e ciò è dovuto ad un processo di invecchiamento conosciuto come arteriosclerosi. Tale inspessimento può causare una parziale occlusione, con una progressiva e graduale diminuzione del flusso di sangue che arriva alle strutture dell’orecchio interno. I meccanismi dell’equilibrio in genere riescono a superare tali problemi adattandosi, talvolta però si può avere una instabilità persistente. Tutto ciò può peggiorare per gli improvvisi cambiamenti di posizione come avviene quando ci si alza dal letto e si gira la testa improvvisamente. Una completa occlusione dei vasi dell’orecchio interno (trombosi), produce una vertigine acuta spesso accompagnata da vomito e nausea. I sintomi possono continuare per alcuni giorni, con una graduale diminuzione delle vertigini in un periodo che varia da alcune settimane ad alcuni mesi, quando cioè l’orecchio controlaterale sostituisce le funzioni dell’orecchio malato.
· c) Emorragia: Raramente uno dei piccoli vasi dell’apparato dell’equilibrio si può rompere. Ciò può avvenire spontaneamente, per una ragione sconosciuta, o può essere la conseguenza della ipertensione, o di un trauma cranico. I sintomi sono sovrapponibili a quelli conseguenza di una stenosi del vaso. Trattamento Il trattamento della vertigine dovuta a disturbi della circolazione consiste in una terapia medica a base di anti-vertiginosi, di preparati che migliorano la circolazione (vasodilatatori), e sedativi. Un individuo con tale tipo di vertigine dovrebbe evitare le droghe che danno vasocostrizione vascolare come la caffeina (caffè , thè) e la nicotina (sigarette). Gli stress emotivi, l’insonnia, e la eccessiva fatica fisica o intellettuale, dovrebbero essere evitati il più possibile.
Vascolarizzazione dell’orecchio interno
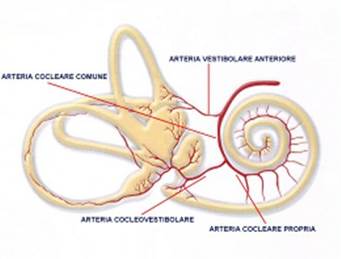
Il labirinto è irrorato dall’arteria uditiva interna (AUI, o arteria labirintica), vaso che nasce più spesso dall’arteria cerebellare antero-inferiore, più raramente dall’arteria basilare o dall’arteria cerebellare superiore. Entrata nell’orecchio interno dopo aver percorso il CUI, l’AUI si divide in due rami principali: l’arteria cocleare comune e l’arteria vestibolare anteriore. L’arteria cocleare comune si divide in nell’arteria cocleare propriamente detta (o arteria cocleare principale) e nell’arteria vestibolo-cocleare. La prima forma un plesso all’interno della coclea irrorando il ganglio spirale, le strutture della membrana basilare e la stria vascolare. La seconda, a sua volta, dà un ramo cocleare (per il giro basale della chiocciola) e un ramo vestibolare (definita arteria vestibolare posteriore) che garantisce l’apporto vascolare alla parte inferiore del sacculo e all’ampolla del canale semicircolare posteriore (CSP).
L’altro ramo principale dell’AUI, l’arteria vestibolare anteriore, fornisce rami per l’utricolo, per il canale semicircolare anteriore (CSA), per il canale semicircolare laterale (CSL) e per una piccola parte del sacculo (Figura 4) (Mazzoni, 1990). E comprensibile quindi come le varie parti del labirinto possono essere selettivamente colpite da un evento ischemico. Poiché la vascolarizzazione dell’orecchio interno è di tipo terminale, priva cioè di vasi collaterali che possano supplire eventuali deficit di irrorazione, tale organo appare estremamente suscettibile ad eventi ischemici; sono sufficienti 15 secondi di ischemia perché si instauri un danno a carico della trasmissione nervosa, anche se è possibile un recupero fino a 15-30 minuti di distanza da una completa ostruzione del flusso. In caso di ostruzione di più lunga durata il danno risulta tuttavia irreversibile.
|
|
|
|
|
Fig. 2. Irrigazione orecchio interno: 1) arteria cerebellare anteroinferiore (AICA);2)arteria auditiva interna (labiríntica); 3) arteria cocleare comune; 4) artéria vestibolare anteriore; 5) arteria cocleare; 6) arteria cocleo – vestibolare; 7) ramo cocleare; 8) arteria sacculare; 9) arteria ampollare posteriore (GONZALES et al, 1968). |
|
|
Sistema venoso dell’orecchio interno Schuknecht 1986..
|



Il drenaggio venoso del labirinto risulta oltremodo variabile (Mazzoni, 1990). Utricolo ed ampolla dei canali semicircolari laterale e anteriore fanno capo alla vena vestibolare anteriore. Il sacculo, il canale semicircolare posteriore ed il giro basale della chiocciola fanno capo alla vena vestibolare posteriore. Queste due vene confluiscono nella vena della finestra rotonda formando la vena vestibolococleare che, unendosi alla vena modiolare proveniente dalla coclea, forma la vena dell’acquedotto della chiocciola, che sbocca poi nel seno petroso inferiore. Alcuni rami venosi provenienti dai canali semicircolarì conflui scono nella vena dell’acquedotto del vestibolo, che scorre lungo il dotto endolinfatico per aprirsi nel seno laterale (Figura .5).
Caratteristiche Della Microcircolazione Cocleovestibolare
J.-P. Sauvage, S. Puyraud, N. Khalifa. Sordità improvvise e fluttuanti . EMC – Otorinolaringoiatria 2005:1-16 [Article 20-183-A-10].
La vascolarizzazione dell’orecchio interno è di tipo terminale e ogni rallentamento o qualsiasi interruzione del flusso ematico cocleare (FEC) provoca un’anossia cocleare. È noto da molto tempo che l’occlusione sperimentale dell’arteria labirintica deteriora molto rapidamente e definitivamente la coclea[ Martin et al.,2000; Mom et al.,1999 ] in meno di 1 ora. Le cellule ciliate esterne sembrano più vulnerabili delle cellule ciliate interne. Teoricamente, una terapia effettuata oltre 1 ora dopo l’esordio dell’ischemia non ha alcun opportunità di successo. L’organo del Corti degenera con in seguito comparsa di fibrosi e ossificazione. In tali condizioni, come spiegare che una sordità improvvisa di probabile origine vascolare possa recuperare.
Alcuni studi su un modello animale di ischemia cocleare reversibile e selettiva hanno mostrato che la coclea poteva tollerare ischemie complete di oltre 5 minuti e recuperare dopo un periodo di disfunzione transitoria. [ Mom et al.,2003 ] Inoltre, anche se la vascolarizzazione della coclea dipende esclusivamente dall’arteria labirintica, essa possiede anche un dispositivo longitudinale alimentato da un dispositivo raggiato ripartito che permette la regolarizzazione dell’apporto ematico ai diversi settori della coclea. Occlusioni selettive dei rami di divisione dell’arteria labirintica provocano soltanto dei deficit uditivi parziali e reversibili. [ Ueda et al.,1998 ]
Infine, il modo in cui la sordità recupera dopo un accidente vascolare parziale che interessi la coclea dipende dalla disposizione dei vasi principali e dal modo in cui si ridistribuisce il circolo intracocleare in funzione della sede dell’interruzione (Fig. 1) . La disposizione delle arterie cocleari è in effetti molto variabile. [ Mazzoni A. 1972 ] L’arteria labirintica può essere doppia nel 50% dei casi. L’arteria cocleare propria può essere assente e quindi l’arteria cocleovestibolare irrora da sola tutta la coclea. L’arteria labirintica può nascere da una grossa arteria cerebellare antero-inferiore o direttamente dal tronco basilare. Viceversa, può nascere da una sottile arteria cerebellare postero-inferiore o direttamente da un’arteria vertebrale ipoplasica. Tenere in considerazione questi fattori anatomici permette così di prevedere in qualche modo e allo stesso tempo il tipo audiometrico riscontrato, la possibile associazione a vertigini e la capacità di recupero (Tabella 1) .
|
Tabella 1 – Sintomi e possibilità di recupero teoriche in caso di occlusione ipotetica dei vasi cocleari, secondo Hultcrantz 1999 modificato. |
||||
|
Vaso interessato |
Variante |
Caratteri della sordità |
sintomi vestibolari |
Recupero |
|
Arteria labirintica |
Unica |
Sordità totale |
Forte vertigine prolungata |
Molto compromesso |
|
Doppia |
Interessamento delle basse e medie frequenze o curva piatta |
Vertigini di qualche ora per la conservazione del sacculo e dell’ampolla del canale posteriore |
Possibile |
|
|
Arteria labirintica a valle dell’origine dell’arteria vestibolare anteriore |
Unica |
Sordità grave su tutte le frequenze |
Vertigini per interessamento del sacculo e dell’ampolla posteriore |
Cattivo con possibile irrorazione da rami vestibolari |
|
|
Doppia |
Interessamento frequenze basse o medie o curva piatta |
Nessuna vertigine |
Possibile per la seconda arteria labirintica |
|
Arteria vestibolare anteriore |
– —— |
Nessuna sordità |
Sindrome di Lindsay e Hemenway* |
Possibile con il ramus vestibularis |
|
Arteria vestibolococleare |
– ———- |
Lesione grave degli acuti |
Vertigine per interessamento sacculare e del canale posteriore |
Possibile per arteria cocleare propria (III) |
|
Nessuna arteria cocleare propria |
Sordità grave su tutte le frequenze |
Idem |
Molto compromesso |
|
|
Arteria cocleare propria |
– ———- |
Interessamento delle frequenze medie e basse |
Nessuna vertigine |
Possibile per l’arteria vestibolococleare e il ramus cochlearis |
|
|
o curva piatta |
|
|
|
|
Ramus vestibularis |
– ———- |
Nessuna sordità |
Vertigine per interessamento del sacculo e del canale semicircolare posteriore |
Buono per l’arteria vestibolare anteriore |
|
Ramus cochlearis |
– ——— |
Perdita temporanea degli acuti |
Nessuno |
Buono poiché l’occlusione è situata tra II e III: supplenza da parte de flusso del legamento spirale |
|
Arteria spirale modiolare |
– ——— |
Interessamento delle frequenze medie e basse più grave che per l’arteria cocleare propria |
|
|
|
Ossificazione secondaria |
||||
|
Arteriole radiali |
– ——- |
Nessun segno clinico, lesione troppo piccola, circolo collaterale dal flusso del legamento spirale |
|
|
|
Vena dell’acquedotto cocleare |
– ———- |
Perdita su tutte le frequenze, ma meno rilevante che in caso di lesione arteriosa |
|
|
|
Vene dell’acquedotto vestibolare |
– ———- |
Nessuna sordità |
Vertigini per interessamento di tutte le strutture vestibolari |
|
|
*Sindrome di Lindsay e Hemenway: grande vertigine ad esordio improvviso e regressione progressiva, con areflessia calorica unilaterale, immediatamente seguito da vertigine posizionale parossistica benigna che coinvolge il canale semicircolare posteriore omolaterale al lato areflessico. |
||||
Altri fattori sono da tenere presenti: la grande sensibilità ai traumi sonori nel periodo peri-ischemico [ Mom et al.,1999 ] e l’età che costituisce un fattore di devascolarizzazione con riduzione del FEC di circa il 20%. [ Booth JB 1997 ]
Aspetti fisiopatologici del circolo labirintico (Dr. A Castiglione Anno Accademico 2010/11 -Tesi di Specializzazione in Audiologia e Foniatria Dir. Prof. Stefano Pelucchi) POACUSIA IMPROVVISA:RICERCA DI NUOVI MARKERS MOLECOLARI
Come già accennato in precedenza la particolare vascolarizzazione del labirinto interno induce molte riflessione sulla genesi vascolare di alcune malattie, coinvolgendo non solo la parete dei vasi (contenitore), ma anche il suo contenuto (globuli rossi, proteine, ioni, metaboliti tossici). Queste acquisizioni hanno di fatto spostato l’interesse dal macrocircolo al microcircolo e hanno spinto a paragonare la vascolarizzazione cocleare ad un altro organo molto importante: il rene(8,9) (Fig. 8). In tale contesto appare fondamentale il ruolo svolto dall’organo endoteliale. L’endotelio non è infatti una semplice barriera tra il sangue e la parte extracellulare, ma è un organo estremamente vitale e dinamico. L’endotelio è l’organo che bilancia i fattori pro-aggreganti con quelli antiaggreganti (bilancia emostatica), produce monossido d’azoto (NO) ad azione vasodilatante e subisce un’azione di vasocostrizione ad opera dell’angiotensina II oppure dell’endotelina. Nella struttura endoteliale vi è inoltre una riserva d’interleuchine pro-infiammatorie che si liberano sotto l’azione di fattori di rischio vascolari, meccanici, metabolici (iperglicemia, alterazioni ormonali) e trombotici.
![]() I fattori di rischio vascolari possono di fatto influenzare la funzionalità endoteliale: stasi, iperglicemia, iperlipidemia, determinano perdita di produzione del monossido
I fattori di rischio vascolari possono di fatto influenzare la funzionalità endoteliale: stasi, iperglicemia, iperlipidemia, determinano perdita di produzione del monossido
|
|
|
Fig. 3A – Rappresentazione schematica dell’irrorazione arteriosa cocleo-vestibolare, normale, di un orecchio destro. L’arteria uditiva interna è un ramo dell’arteria basilare o della cerebellare inferiore e attraversa il meato acustico interno insieme al pacchetto stato-acustico-facciale le strutture dell’orecchio in-terno (coclea e vestibolo). Qui sfiocca in due rami terminali: arteria vestibolare anteriore ed arteria cocleare comune che a sua volta si divide in a. cocleovestibo-lare e a. cocleare propria.
|
|
|
|
Fig. 3B – L’occlusione dell’arteria cocleare propria, porta ad ipossia/ischemia della coclea (area nera nella figura) con severa ipoacusia ad insorgenza improvvisa per i toni medio-gravi. Solitamente in tale situazione non sono presenti sintomi vestibolari poiché l’irrorazione è garantita da altri rami; possono insorgere acufeni
|
|
|
|
Fig. 3C – L’occlusione dell’arteria cocleo-vestibolare :.Il deficit ematico interessa la porzione basale della coclea ed le macule dell’utricolo e del sacculo è caratterizzata da sintomi vestibolari, uditivi (soprattutto a carico delle alte frequenze) e acufeni. Compare un nistagmo di I grado che batte verso il lato sano e deviazioni (nella marcia o degli arti) verso il lato leso).La sintomatologia vestibolare legata alla patologia maculare quale l’ocular tilt reaction. |
|
|
|
Fig. 3D – L’occlusione dell’arteria cocleare comune comporta l’anacusia e sintomi vestibolari. Il danno interessa contemporaneamente sia la colclea nella sua totalità che le macule utriculo-sacculari. la sintomatologia è caratterizzata sia dall’anacusia che dal danno maculare con la ocular tilt reaction. |
|
|
|
Fig. 3E – In caso di ostruzione dell’arteria vestibolare anteriore è l’intero apparato vestibolare ad essere interessato con risparmio del sacculo e della coclea. La sintomatologia vestibolare risulta particolarmente intensa caratterizzato della sindrome di Lindsay-Hemenway: vertigini intense che si accentuano con i cambiamenti del capo e vomito intenso. |
|
|
|
Fig. 3F – Questa è la situazione peggiore con danno esteso alla coclea ed al vestibolo: ne conseguono l’anacusia e sintomi neurovegetativi e vestibolari intensi. |





I fattori di rischio vascolari possono di fatto influenzare la funzionalità endoteliale: stasi, iperglicemia, iperlipidemia, determinano perdita di produzione del monossido d’azoto e conseguente riduzione della vasodilatazione capillare. La produzione di NO appare fondamentale per l’emodinamica dei distretti vascolari e la sua regolazione sembra essere legata alle proteine che trasmettono alla parete vascolare i segnali meccanici (pressione) chimici (es. glicemia) e biologici (es. enzimi) che circolano nel flusso ematico.
Fig. 4 – Gli aspetti del microcircolo cocleare hanno indotti molti autori a paragonarlo al nefrone. A) Immagine microscopica della vascolarizzazione cocleare; B) schema della distribuzione dei vasi nei giri cocleari; C) rappresentazione schematica della distribuzione delle pompe ioniche lungo il nefrone e nella stria vascolare
Infine periciti e miociti appaiono dotati di granulazioni citoplasmatiche ricche di istamina, serotonina, chinine e soprattutto prostaglandine, tutte sostanze ad azione vasoattiva. Si determina così una fine modulazione che può adeguare, momento per momento, la risposta vascolare alle esigenze metaboliche locali (autoregolazione del microcircolo), come avviene ad esempio, in situazioni di stress funzionale (esposizione a rumore) quando la perfusione cocleare sembra aumentata.
La stria vascolare. I meccanismi fisiologici dell’udito richiedono un notevole apporto di energia e ossigeno, garantiti continuamente dalla stria vascolare. Questa inoltre contribuisce in maniera essenziale alla produzione di endolinfa e al mantenimento delle sue caratteristiche elettrochimiche, indispensabili al corretto funzionamento delle cellule ciliate e alla generazione dei potenziali endococleari. La stria vascolare è situata sulla superficie interna del legamento spirale e si estende dalla prominenza spirale fino alla membrana di Reissner. È sostanzialmente un epitelio riccamente vascolarizzato, la cui sezione mostra infatti numerosi capillari e tre tipi di cellule, superficiali e altamente specializzate:
– le cellule marginali;
– le cellule intermedie;
– le cellule basali.
I capillari della stria vascolare hanno una direzione prevalentemente longitudinale e sono caratterizzati da pareti sottili, senza periciti, con spazi pericapillari poco sviluppati; formano alla base della coclea una rete a maglie molto spesse che si semplifica andando verso l’apice.
|
|

Il lume di questi capillari è notevole, caratterizzato da un’alta densità di globuli rossi; ne consegue una velocità circolatoria molto bassa con un verosimile tasso di scambio metabolico molto elevato. La permeabilità capillare a questo livello contrasta con l’impermeabilità della maggior parte dei capillari del labirinto cocleovestibolare, i quali sembrano formare una sorta di barriera ematolabirintica, simile a quella ematoencefalica. Questa permeabilità può essere aumentata o diminuita in caso di ipertensione o ipotensione vascolare indotte sperimentalmente. A livello della stria vascolare, la permeabilità capillare è regolata dagli spazi endo- e peri-linfatici attraverso giunzioni serrate intercellulari. Le cellule marginali della stria vascolare formano il primo strato di cellule (dal lume verso l’interno) e sono tutte legate tra loro, nello spazio intercellulare superiore, da giunzioni serrate; sono le uniche cellule epiteliali della stria vascolare e ricche di cito cheratine.
Le cellule intermedie (lo strato compreso tra le marginali e le basali) sono situate nella parte mediana della stria vascolare, non raggiungono il lume, ma inviano delle digitazioni tra le cellule marginali; sono di origine mesenchimale, ricche
in proteine filamentose tipo la vimentina. Le cellule intermedie poggiano direttamente sui capillari mediante processi dendritici ramificati. Sarebbero veri e propri melanociti appartenenti al sistema APUD, vale a dire con funzioni paracrine/endocrine e liberazione di neurotrasmettitori ad azione locale in grado di influenzare le secrezioni cellulari, il flusso ematico e la contrazione di cellule muscolari lisce.
Le cellule basali sono a contatto con il legamento spirale imbevuto di perilinfa e somigliano ai fibrociti del legamento spirale (Fig. 5).
Parametri del microcircolo cocleare. I principali parametri del microcircolo sono: emodinamica, reologia, ematocriti locali, viscosità ematica capillare apparente e vasomotilità.
L’emodinamica del microcircolo della stria vascolare è caratterizzata da un gradiente pressorio con riduzione massima a livello delle arteriole distali. La reologia è condizionata dall’effetto Fahraeus-Lindqvist: i globuli rossi si dispongono in fila al centro, quando raggiungono capillari inferiori a 1 mm, lasciando alla periferia del lume un “manicotto” plasmatico. Questo fenomeno permette un aumento della velocità dei globuli rossi con una riduzione apparente della viscosità; tuttavia nei capillari di diametro inferiore (5 mm) il fenomeno risulta esattamente invertito, la viscosità ematica apparentemente aumenta e la velocità dei globuli rossi si riduce.
A livello del microcircolo cocleare la concentrazione dei globuli rossi non è uniforme nei vari distretti. Nella stria vascolare ad esempio, poiché il vaso a diametro maggiore “sequestra” la maggior parte dei globuli rossi che sopraggiungono, le arteriole più piccole possono risultare perfuse solo da plasma; questo fenomeno è detto di separazione plasmatica.
Come già detto la viscosità ematica capillare apparente dipende dal tipo (deformabilità), dal numero e dalla disposizione degli elementi cellulari. I leucociti determinano un aumento notevole della viscosità; questa condizione è particolarmente importante durante episodi di leucocitosi, situazione che può quindi predisporre ad importanti e dannose alterazioni del microcircolo cocleare.
La vasomotilità delle arteriole distali influisce direttamente sul numero dei capillari perfusi e sulla ripartizione locale del sangue, soprattutto durante aumento di richieste metaboliche da parte di un distretto. Diversi studi sono stati condotti a tale riguardo e da questi è risultato che la simpatectomia influisce significativamente sul calibro dei capillari della stria vascolare. Ma secondo altri autori l’impatto funzionale reale degli agonisti e antagonisti vasomotori resta discutibile e difficilmente accertabile(6).
Accoppiamento fibro-vascolare nella stria vascolare.

Fig. 6
Per comprendere come l’orecchio possa regolare il proprio flusso sanguigno, sono stati condotti numerosi studi sulla capacità dei capillari della stria vascolare di rispondere a stimoli e segnali in diverse situazioni. Uno di questi ha evidenziato una stretta relazione tra fibrociti e capillari che consentirebbe un aumento del flusso locale in presenza di calcio e liberazione di ossido nitrico. Nella figura 10 è riportato una schema riassuntivo dell’accoppiamento fibro-vascolare in grado di rispondere all’aumentare delle esigenze metaboliche della coclea in presenza di uno stimolo acustico(10). Fig. 6
Patologie associate ad alterazioni del flusso ematico
Oltre all’ipoacusia improvvisa, di cui abbiamo già parlato, sono molte le patologie dell’orecchio interno che riconoscono una base vascolare; qui di seguito vengono analizzate le principali con alcune considerazioni generali Nakashima et al 2003(11).
Noise-Induced Hearing Loss. Tra i diversi meccanismi fisiopatologici che sostengono l’ipoacusia transitoria o permanente da esposizione al rumore, bisogna sicuramente includere la riduzione del flusso sanguigno cocleare; diversi studi hanno dimostrato che l’esposizione al rumore comporta una vasocostrizione dei capillari della membrana basilare, del legamento spirale e della stria vascolare. È stato inoltre accertato un aumento della pressione arteriosa sia nell’uomo sia nell’animale, accompagnata da alterazioni del metabolismo del magnesio. Il magnesio sembra volgere un ruolo protettivo nei confronti del rumore. Un’esposizione prolungata a suoni non confortevoli può aumentare notevolmente la produzione di radicali liberi, contribuendo alla patogenesi dell’ipoacusia da rumore. Da queste considerazioni iniziali, si è ipotizzato che la tipica lesione nel profilo audiometrico a 4-6 kHz sia attribuibile all’area di massima sollecitazione della membrana basilare, con conseguente massima riduzione del flusso locale.
Idrope endolinfatica. Sebbene molti autori non abbiano riscontrato differenze significative nel flusso sanguigno di coclee con idrope rispetto a coclee normali, è stata accertata un’alterazione dell’autoregolazione locale: l’idrope endolinfatica riduce il flusso sanguigno cocleare.
Presbiacusia. Che all’invecchiamento possano associarsi alterazioni del flusso locale e sistemico è risaputo; tuttavia non sono chiare le relazioni temporali e causali che portano all’ipoacusia e alla degenerazione delle strutture vascolari.
Acquedotto vestibolare allargato. Il rapporto tra perfusione cocleare e pressione dei fluidi cocleari è identico a quello esistente tra perfusione cerebrale e liquido cerebrospinale, per cui all’aumentare della pressione dei fluidi si ha una riduzione del flusso sanguigno. L’acquedotto vestibolare o cocleare allargati rappresentano condizioni patologiche con aumento della pressione dei fluidi dell’orecchio interno; la conseguenza di ciò è una riduzione del flusso sanguigno cocleare. D’altro canto, anche in situazioni opposte si può avere ipoacusia: una drastica e rapida riduzione della pressione dei fluidi può comportare danno della funzione uditiva anche in presenza di un adeguato flusso arterioso.
Emopatie. Come già accennato, tutte quelle condizioni che aumentano la viscosità ematica possono comportare un danno da ipoperfusione cocleare: leucemia, leucocitosi, policitemia, anemia falciforme, crioglobulinemia e macroglobulinemia sono tutte condizioni che possono alterare il flusso sanguigno cocleare e l’ipoacusia può essere il primo sintomo della malattia. Queste patologie inoltre possono portare ad una fibrosi degli spazi perilinfatici e causare episodi di microembolismo.
Disordini dei vasi sanguigni. Dopo quanto riportato finora, non ci si stupirà del fatto che tutti i disordini dei vasi, dalle vasculiti autoimmuni alle sindromi congenite, possono avere ripercussioni importanti sull’organo dell’udito. Anche il diabete com’è noto, soprattutto negli stadi più avanzati, può portare ad un danno notevole del microcircolo di tutti i distretti anche di quello cocleare, con perdita della capacità di autoregolazione ed ispessimento delle pareti dei vasi Kariya et al2010(12).
Tra le altre patologie che riconoscono una base vascolare nell’insorgenza dell’ipoacusia vale la pena ricordare la Susac Syndrome, la Norrie Syndrome e l’arterite di Takayasu.
Occlusione del drenaggio venoso. Sebbene sia lecito pensare che un’occlusione del drenaggio venoso possa influire negativamente sul flusso sanguigno coclea-re, studi condotti sugli animali non hanno dimostrato una significativa associazione tra occlusione delle vene dell’orecchio interno ed insorgenza di ipoacusia.
Ruolo dell’ossido nitrico (NO) o ossido di azoto o mosossido di azoto. L’ossido di azoto ha un ruolo determinante nella regolazione del flusso cocleare; diversi studi hanno dimostrato che l’ossido di azoto è in grado di aumentare il flusso sanguigno cocleare e proteggere la coclea in alcune condizioni di stress; la produzione di NO risulta aumentata, dalla presenza di ossidositentasi inducibile, durante l’ischemia, l’ototossicità e l’idrope endolinfatica. Oltre a svolgere un ruolo determinante nel regolare il flusso sanguigno cocleare, NO sembra avere anche un ruolo di neurostrasmettitore.
L’ossido nitrico è un gas incolore molto reattivo perché di fatto è un radicale libero: la sua molecola (NO) è formata da un atomo di azoto legato ad un atomo di ossigeno. Quando agisce da messaggero NO viene prodotto in continuazione a bassi livelli dalle cellule e controlla la contrazione delle cellule muscolari e la crescita delle cellule nervose. L’ossido nitrico è particolarmente efficace come messaggero: diffonde rapidamente perché la sua molecola è molto piccola e apolare, ma rimane abbastanza localizzato perché è molto reattivo e viene distrutto rapidamente. L’ossido nitrico può anche agire come sostanza tossica grazie alla sua grande reattività. I macrofagi, cellule del sistema immunitario, lo utilizzano per uccidere i patogeni insieme con altri composti reattivi dell’ossigeno.
Le cellule producono tre tipi di ossido nitrico sintasi (NOS) che generano ossido nitrico per le diverse funzioni. La NOS neuronale e la NOS endoteliale producono continuamente bassi livelli di NO che agisce rispettivamente come neurotrasmettitore e come vasodilatatore. La NOS inducibile, invece, produce maggiori quantità di NO che risultano tossiche e servono per combattere i patogeni.
Tutti e tre questi enzimi sono complessi e sono composti di molte subunità che svolgono funzioni diverse. I ricercatori sono riusciti a determinarne la struttura scomponendoli nelle singole parti.
La subunità ossidante produce NO con l’aiuto di un gruppo eme che aggiunge un atomo di ossigeno, proveniente da O2, all’azoto nella catena laterale di un amminoacido di arginina (gialla) che viene trasformata in citrullina. Questa è stata la prima subunità ad essere studiata con la cristallografia ed è stata determinata dapprima su una NOS inducibile e in un secondo momento su una NOS neuronale.
La subunità riducente dona elettroni a quella ossidante e contiene coenzimi riducenti come NADPH, FAD e FMN.
Il breve segmento che unisce le due subunità è legato alla calmodulina che controlla il flusso di elettroni.
Nel caso della NOS endoteliale, il messaggio portato da NO è ricevuto dall’enzima guanilato ciclasi solubile, un enzima complesso che inizia una catena di eventi a cascata all’interno della cellula. Quando si lega ad una molecola di NO, l’enzima si attiva e trasforma GTP in GTP ciclico (cGTP). Questa molecola agisce quindi come secondo messaggero attivando delle chinasi che a loro volta fosforilano la miosina provocando il rilassamento delle cellule muscolari.
 Come NOS, anche la guanilato ciclasi solubile è un enzima complesso formato da molti domini che è stato studiato per parti dai cristallografi. La porzione che lega NO è in alto dove si vede un gruppo eme che lega l’ossido nitrico. La porzione ciclasi è mostrata in basso e vi sono poi molti altri domini che legano queste due parti. Nella figura qui sotto è mostrato il dettaglio del legame tra NO ed eme. L’ossido nitrico (azoto azzurro e ossigeno rosso) si lega obliquamente all’atomo di ferro (arancione) che si trova al centro dell’eme. La catena proteica è mostrata in viola (Fig. 7).
Come NOS, anche la guanilato ciclasi solubile è un enzima complesso formato da molti domini che è stato studiato per parti dai cristallografi. La porzione che lega NO è in alto dove si vede un gruppo eme che lega l’ossido nitrico. La porzione ciclasi è mostrata in basso e vi sono poi molti altri domini che legano queste due parti. Nella figura qui sotto è mostrato il dettaglio del legame tra NO ed eme. L’ossido nitrico (azoto azzurro e ossigeno rosso) si lega obliquamente all’atomo di ferro (arancione) che si trova al centro dell’eme. La catena proteica è mostrata in viola (Fig. 7).

Fig. 7 – L’ossido nitrico è fortemente legato ad un gruppo eme, che contiene un atomo di ferro (in arancione). Il messaggio portato da NO è ricevuto dall’enzima guanilato ciclasi solubile. Questo enzima inizia una serie di eventi a cascata all’interno della cellula.
Le tre isoforme di NOS sono molto simili e quindi i ricercatori stanno cercando di sfruttare alcune piccole differenze per creare un farmaco che ne blocchi una senza influenzare le altre. Questo sarà molto utile per curare alcune malattie, per esempio la iNOS, ossido nitrico sintasi inducibile, ha un ruolo chiave nello sviluppo del morbo di Parkinson e in quello di Alzheimer e anche nella sclerosi multipla, quindi farmaci in grado di bloccare la iNOS, ma non le altre due, potrebbero aiutare nel trattamento di queste malattie.
Purtroppo, però, i siti attivi delle tre isoforme di NOS sono praticamente identici e quindi i ricercatori stanno sintetizzando dei farmaci più grandi che arrivino ad
interagire anche con altri punti dell’enzima dove si manifesta una differenza sfruttabile tra le tre isoforme.
Ruolo degli ioni ferroso (Fe2+, forma ridotta) e ferrico (Fe3+, forma ossidata).
In questo paragrafo cercherò d’illustrare il ruolo degli ioni del ferro, sottolineando l’importanza di una corretta omeostasi intra ed extracellulare volta a garantire due processi fondamentali: sintesi di proteine e processi infiammatori locali. Da quando la teoria dei radicali liberi si è largamente diffusa, numerosi sono stati gli studi che hanno dimostrato come la contemporanea presenza di ioni del ferro a livello cocleare potesse aggravare l’azione degenerativa dei ROS, fino a favorirne la formazione. Inoltre concentrazioni elevate di ioni ferro sono di per sé tossiche ed in grado di avviare processi apoptotici e pro-infiammatori con richiamo di cellule del sistema reticolo-endoteliale. Inutile sottolineare d’altro canto quanto stati carenziali possano avere ripercussioni serie sulla sintesi di proteine. È pure noto che stati di emocromatosi possono arrivare a danneggiare interi organi. Si cercherà qui di discutere però solo di quei fini meccanismi di equilibrio omeostatico non direttamente correlabili a franchi stati carenziali o di accumulo, tentando di elucidare le tappe biochimiche, che se alterate o disturbate nella loro funzione, possono avere importanti ripercussioni fisiopatologiche. Il punto di partenza è senz’altro il lavoro di Philine Wangemann del 2009 “Cochlear Homeostasis and Homeostatic Disorders” un’importante review sui fini meccanismi di regolazione dell’omeostasi cocleare. Il primo importante paragrafo è dedicato a quelle che secondo Wangemann sono i processi biochimici più importanti e delicati a livello cocleare: la produzione di energia (vedi figura 8) e l’omeostasi ossidoriduttiva (vedi figura 9). Cioè Wangemann equipara l’importanza della disponibilità di energia, a tutti evidente, a quella della capacità di smaltimento dei radicali liberi.

![]() Questi processi sono localizzati soprattutto a livello della stria vascolare confermando il suo ruolo chiave. Non sarà a tal proposito un caso che i principali depositi di ferro siano proprio nella stria vascolare come confermato dagli studi di Hinojosa del 1972 Acta Otolaryngol. 1972 Jul-Aug;74(1):1-14, di Santos-Sacchi del 1985 Acta Otolaryngol. 1985 Jul-Aug; 100(1-2):26-32, e di Kakigi del 2011 Otol Neurotol. 2011 Jul;32(5):856-62.
Questi processi sono localizzati soprattutto a livello della stria vascolare confermando il suo ruolo chiave. Non sarà a tal proposito un caso che i principali depositi di ferro siano proprio nella stria vascolare come confermato dagli studi di Hinojosa del 1972 Acta Otolaryngol. 1972 Jul-Aug;74(1):1-14, di Santos-Sacchi del 1985 Acta Otolaryngol. 1985 Jul-Aug; 100(1-2):26-32, e di Kakigi del 2011 Otol Neurotol. 2011 Jul;32(5):856-62.
I radicali liberi, generati in quantità controllate, possono servire come un segnale molecolare e fanno parte della omeostasi cellulare redox. La persistenza di radicali liberi, tuttavia, comporta uno squilibrio ossidoriduttivo con ossidazione incontrollata di proteine e lipidi, e danno o morte cellulare. I radicali liberi sono un sottoprodotto di un inefficiente trasferimento elettronico nella catena di trasporto mitocondriale e nel sistema mitocondriale del cit. P450. L’incompleta riduzione di O2 genera il radicale libero •O2– (anione superossido), il quale comunque può formarsi anche da altre vie metaboliche. •O2– viene convertito a
H2O2 (perossido di idrogeno) e O2 in presenza di H+ (vedi figura 9). Il perossido di idrogeno, in presenza di ferro dà luogo alla formazione di un radicale estremamente aggressivo, il gruppo idrossile •OH– in quella che è conosciuta come la reazione di Fenton. In alternativa, •O2 – (anione superossido) può reagire con l’ossido di azoto radicale •NO– a formare il perossinitrato ONOO– che, in condizioni acide o in presenza di CO2, attraverso la formazione di NO2– provoca nitrazione delle proteine, dei lipidi e degli acidi nucleici.
 |
In conclusione, il ferro nell’eme è necessario per il trasporto, il legame, e il rilascio di ossigeno; la pronta disponibilità di ferro, per essere incorporato in qualunque gruppo eme, è essenziale per la sopravvivenza degli organismi. Il ferro è quindi indispensabile per la funzione di enzimi coinvolti in numerosi processi cellulari critici. Tuttavia, il ferro dona elettroni per la generazione del radicale superossido, e può partecipare alla generazione di radicali idrossilici tramite la reazione di Fenton (vedi figura 10). La tossicità del ferro in sistemi cellulari è attribuibile in gran parte alla sua capacità di partecipare alla generazione di tali specie reattive che possono danneggiare direttamente il DNA, lipidi, proteine, portando a profonda tossicità cellulare. Per tanto, il bilanciamento del ferro è mantenuto con cura raffinata. La ferritina, catturando e regolando la quota disponibile intracellulare di ferro “labile”, svolge un ruolo chiave nel mantenimento dell’omeostasi (Regulation of ferritin genes and protein, 2002 Frank M. Torti and Suzy V. Torti).
Sistemi di detossificazione. È interessente notare però che gli ioni ferro, e in genere tutti i metalli coinvolti nell’equilibrio redox, sono direttamente coinvolti anche nelle vie di detossificazione dei radicali liberi come ad esempio quelle regolate dalla SOD (superossido dismutasi) e rappresentate dalle seguenti reazioni:
M(n+1)+ − SOD + O2 − → Mn+ − SOD + O2 ; Mn+ − SOD + O2− + 2H+ → M(n+1)+ − SOD + H2O2 ; dove M = Cu (n=1) ; Mn (n=2) ; Fe (n=2) ; Ni (n=2).
Esistono molte forme comuni di SOD: sono proteine che possono avere cofattori metallici diversi, come rame, zinco, manganese, ferro o nichel. Nell’uomo, sono presenti tre forme di superossido dismutasi. La SOD1 si trova nel citoplasma, la SOD2 nei mitocondri mentre la SOD3 è extracellulare. La prima è un dimero (consiste di due unità), mentre le altre sono tetrameri (quattro subunità). La SOD1 e la SOD3 contengono rame e zinco, mentre la SOD2 ha la manganese nel suo centro di reazione. I geni sono collocati nei cromosomi 21, 6 e 4, rispettivamente (21q22.1, 6q25.3 and 4p15.3-p15.1).
Ferroportina e iNOS. Abbiamo visto il ruolo del ferro, dell’NO e dell’iNOS; alla luce di quanto detto vediamo ora di aggiungere un altro elemento e capire come inserirlo in questa complessa catena di eventi. Il ferro è indispensabile alla formazione delle NOS, però in alcuni studi è stato dimostrato che paradossalmente, la presenza di ferro in eccesso nelle cellule inibisce la sintesi di iNOS(13) (ossido sintetasi inducibile) proprio quella più importante nelle condizioni di stress (Fig. 10). Ne consegue che mutazioni della ferroportina, trasportatore del ferro, siano indirettamente in grado di inibire la sintesi di iNOS e che la presenza intracellulare di ferro potenzi lo stress ossidoriduttivo.Il regolatore centrale del metabolismo del ferro è l’epcidina (HEPC), un peptide antimicrobico circolante che è prodotto dal fegato in risposta al ferro ed a segnali infiammatori. L’epcidina sierica interagisce con la proteina che esporta il ferro, la ferroportina, espressa sulla superficie di macrofagi ricchi di ferro e cellule intestinali. Di conseguenza, la ferroportina è internalizzata e degradata, ed il ferro non necessario rimane nella cellula oppure non è assorbito dagli enterociti. Quando il ferro è scarso (ad es. nell’anemia), la trascrizione epatica dell’epcidina è bloccata e più ferro è rilasciato in circolo tramite la ferroportina per fare fronte alle richieste eritroidi. La ferritina è la molecola deputata all’immagazzinamento intracellulare di ferro. La transferrina (TF) è la proteina 

![]() deputata invece al trasporto sierico del ferro.
deputata invece al trasporto sierico del ferro.
La ferroportina è l’unico esportatore del ferro dalle cellule finora identificato. È espressa sulla membrana basolaterale degli enterociti, nei macrofagi, negli astrociti e negli epatociti (Fig. 11). Mutazioni della ferroportina causano accumulo di ferro nel fegato o nei macrofagi reticoloendoteliali. Tuttavia studi specifici sull’espressione tissutale della ferroportina hanno permesso la sua identificazione in altri distretti: ad esempio proprio nella membrana basolaterale del tubulo prossimale del nefrone(14). In conclusione ci sembra possibile che la ferroportina possa regolare indirettamente l’attività della iNOS e della guanilato ciclasi, quindi contribuire alla patogenesi dell’ipoacusia.
I meccanismi attraverso cui mutazioni della ferroportina (FPN-1), ed in generale alterazioni del metabolismo del ferro, possono influire sull’attività cellulare e delle NOS sono tre:
1) il ferro non è disponibile per la sintesi di NOS;
2) la concentrazione di ferro è tale da inibire la sintesi della iNOS;
3) La presenza di ferro aumenta lo stress ossidoriduttivo.
Sebbene sia auspicabile provare lo stato carenziale o di accumulo del ferro per suffragare tali ipotesi, ciò non è sempre possibile perché il quadro clinico può essere talmente sfumato da non mostrare alterazioni ematochimiche oppure lesioni da accumulo di ferro nei tessuti.
Come invece siano strettamente correlate alcune patologie cocleari con l’attività delle NOS è riportato da diversi studi disponibili in letteratura: in uno dei più recenti (Dai et al.2010) è dimostrata una stretta relazione tra attività della iNOS e capacità dell’orecchio interno di riparare il danno vascolare generato da rumore(15). Molti sono anche gli studi che dimostrano come la presenza di ferro sia in grado di aumentare il danno da radicali liberi e come di contro i chelanti del ferro possono svolgere un’azione protettiva.
Per analogia, è interessante notare come gli effetti dell’NO e delle NOS citati a livello cocleare siano ben noti a livello renale e come in tale ambito siano disponibili molti più articoli in letteratura che possono quindi fornire modelli utili alla corretta interpretazione dei dati disponibili invece per l’organo dell’udito.

Fig. 11 – Metabolismo del ferro e principali enzimi coinvolti.
BIBLIOGRAFIA
1) Mudry A, Tange RA. The vascularization of the human cochlea: its his‑
torical background. Acta Otolaryngol Suppl. 2009;(561):3-16.
2) Delas B, Dehesdin D. Anatomia dell’orecchio esterno. EMC (Elsevier
Masson SAS), Otorinolaringoiatria, 20-010-A-10, 2008.
3) Thomassin JM, Dessi P, Danvin JB, Forman C. Anatomia dell’orecchio
medio. EMC (Elsevier Masson SAS), Otorinolaringoiatria, 20-015-A-10, 2008.
4) Sauvage JP, Stéphane Puyraud S, Roche O, Rahman A. Anatomia
dell’orecchio interno. EMC (Elsevier Masson SAS), Otorinolaringoiatria, 20-020-A-10, 2002.
5) Ferrari Lelli G. Comportamento dell’arteria uditiva interna e dei suoi
rami labirintici nell’uomo. Anatomy and Embryology, 1939; 110, 1:48- 80.
6) Mevio E, Richichi M. Le ipoacusie improvvise. Quaderni Monografici di
Aggiornamento A.O.O.I. 2009.
7) Mannini L, Paniccia R, Cecchi E, Alessandrello Liotta A, Leprini E, Berloco
P, Pagnini P, Abbate R, Franco Gensini G, Prisco D. Reduced erythrocyte deformability and hypercoagulability in idiopathic sudden sensorineural hearing loss. Clin Hemorheol Microcirc. 2005;33(1):47-55.
8) Arima T, Uemura T, Yamamoto T. Structural features of the basal lami‑
na in Reissner’s membrane of the guinea pig. Acta Otolaryngol. 1985 Sep-Oct;100(3-4):194-200.
9) Quick CA, Fish A, Brown C. The relationship between cochlea and kid‑
ney. Laryngoscope. 1973 Sep;83(9):1469-82.
10) Dai M, Shi X. Fibro-vascular coupling in the control of cochlear blood
flow. PLoS One. 2011;6(6):e20652.
11) Nakashima T, Naganawa S, Sone M, Tominaga M, Hayashi H, Yamamoto
H, Liu X, Nuttall AL. Disorders of cochlear blood flow. Brain Res Brain Res Rev. 2003 Sep;43(1):17-28.
12) Kariya S, Cureoglu S, Fukushima H, Morita N, Baylan MY, Maeda Y,
Nishizaki K, Paparella MM. Comparing the cochlear spiral modiolar artery in type-1 and type-2 diabetes mellitus: a human temporal bone study. Acta Med Okayama. 2010 Dec;64(6):375-83.
13) Weiss G, Werner-Felmayer G, Werner ER, Grünewald K, Wachter H,
Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. 1994 September 1; 180(3): 969–976.
14) Wolff NA, Liu W, Fenton RA, Lee WK, Thévenod F, Smith CP. Ferroportin
1 is expressed basolaterally in rat kidney proximal tubule cells and iron excess increases its membrane trafficking. J Cell Mol Med. 2011 Feb;15(2):209-19.
15) Dai M, Yang Y, Omelchenko I, Nuttall AL, Kachelmeier A, Xiu R, Shi X.
Bone Marrow Cell Recruitment Mediated by Inducible Nitric Oxide Synthase/Stromal Cell-Derived Factor-1α Signaling Repairs the Acoustically Damaged Cochlear Blood-Labyrinth Barrier. Am J Pathol. 2010 Dec;177(6):3089-99.
16) Chen CY, Emmerling O, Ilgner J, Westhofen M. Idiopathic sudden sen‑
sorineural hearing loss in children. Int J Pediatr Otorhinolaryngol 2005;69:817-821.
17) Cole RR, Jahrsdoerfer R. Sudden hearing loss. Am J Otol 1988;9:211-
215.
18) Nakashima T, Tanabe T, Yanagita N, Wakai K, Ohno Y. Risk factors for
sudden deafness: a case control study. Aurus Nasus Larynx 1997;24:265-270.
19) Zadeh M H, Storper I.S, Spitzer JB. Diagnosis and treatment of sudden‑
onset sensorineural hearing loss: A study of 51 patients. Otolaryngol Head Neck Surg 2003;128:93-98.
20) Kroenenberg J. Vasoactive therapy versus plasebo in the treatment of
sudden hearing loss: A double-blind clinical study. Laryngoscope 1992;102.
21) Matteson EL Use of methotrexate for autoimmune hearing loss. Ann
Otol Rhinol Laryngol 2000;109:710-714.
22) Chen CY, Halpin C, Rauch SD. Oral steroid treatment of sudden senso‑
rineural hearing loss: a ten year retrospective analysis. Otol Neurotol 2003;24:728-733.
23) Weinaug P. Spontaneous remission in sudden deafness. HNO 1984;32:346-351.
24) Mattox D E, Simmons FB. Natural history of sudden sensorineural
hearing loss. Ann Otol Rhinol Laryngol 1977;86:463-480.
25) SnowJB Jr. Sudden deafness. In: Paparella MM, Shumrick DA, eds. Oto‑
laryngology. Philadelphia: W.B. Saunders, 1980:1757-1766.
26) Mattox DE, Lyles CA. Idiopathic sudden sensorineural hearing loss. Am
J Otol 1989; 10:242-247.
27) Psifidis AD, Psillas GK, Daniilidis JCh. Sudden sensorineural hearing loss:
Long-term follow-up results. Otolaryngol Head Neck Surg 2006;134:809-815.
28) Shaia FT, Sheehy JL. Sudden sensorineural hearing impairment: A re‑
port of 1220 cases. Laryngoscope 1976;86:389-398.
29) Laird N, Wilson WR. Predicting recovery from idiopathic sudden hear‑
ing loss. Am J Otolaryngol 1983;4:161-165.
30) Byl FM. Sudden hearing loss: eight years’ experience and suggested
prognostic table. Laryngoscope 1984;94:647-661.
31) Fetterman B, Saunders J, Luxford W. Prognosis and treatment of sud‑
den hearing loss. Am J Otol 1996;17:529-536.
32) Huy PT, Sauvaget E. Idiopathic sudden sensorineural hearing loss is not
an otologic emergency. Otol Neurootol 2005;26:896-902.
33) Aimoni C, Bianchini C, Borin M, Ciorba A, Fellin R, Martini A, Scanelli G,
Volpato S. Diabetes, Cardiovascular Risk Factors and Idiopathic Sudden Sensorineural Hearing Loss: A Case-Control Study. Audiol Neurotol 2010;15:111–115
34) Gemmati D, Federici F, Catozzi L, Gianesini S, Tacconi G, Scapoli GL,
Zamboni P. DNA-array of gene variants in venous leg ulcers: detection of prognostic indicators. J Vasc Surg. 2009 Dec;50(6):1444-51.
35) Zamboni P, Gemmati D. Clinical implications of gene polymorphisms in
venous leg ulcer: a model in tissue injury and reparative process. Thromb Haemost. 2007 Jul;98(1):131-7. Review.
36) Gemmati D, Tognazzo S, Catozzi L, Federici F, De Palma M, Gianesini S,
Scapoli GL, De Mattei M, Liboni A, Zamboni P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J Vasc Surg. 2006 Sep;44(3):554-62.
37) Zamboni P, Izzo M, Tognazzo S, Carandina S, De Palma M, Catozzi L,
Caggiati A, Scapoli G, Gemmati D. The overlapping of local iron overload and HFE mutation in venous leg ulcer pathogenesis. Free Radic Biol Med. 2006 May 15;40(10):1869-73.
38) Zamboni P, Tognazzo S, Izzo M, Pancaldi F, Scapoli GL, Liboni A, Gem‑
mati D. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J Vasc Surg. 2005 Aug;42(2):309-14.
39) Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews
NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005 Mar;1(3):191-200.
40) Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron
transport and the role of transferring. Biochim Biophys Acta. 2012 Mar;1820(3):188-202.
41) Buretić-Tomljanović A, Vraneković J, Rubeša G, Jonovska S, Tomljanović
D, Sendula-Jengić V, Kapović M, Ristić S. HFE mutations and transferrin C1/C2 polymorphism among Croatian patients with schizophrenia and schizoaffective disorder. Mol Biol Rep. 2012 Mar;39(3):2253-8.
42) Babitt JL, Lin HY. The molecular pathogenesis of hereditary hemo‑
chromatosis. Semin Liver Dis. 2011 Aug;31(3):280-92.
43) Parajes S, González-Quintela A, Campos J, Quinteiro C, Domínguez F,
Loidi L. Genetic study of the hepcidin gene (HAMP) promoter and functional analysis of the c.-582A > G variant. BMC Genet. 2010 Dec 10;11:110.
44) Gemmati D, Zeri G, Orioli E, De Gaetano FE, Salvi F, Bartolomei I,
D’Alfonso S, Asselta R, Dall’Osso C, Leone MA, Singh AV, Zamboni P. Iron Gene Polymorphisms are Associated to Disability, Severity and Early Progression in Multiple Sclerosis. BMC Med Gen. 2012 (in press).
45) Molini E, Serafini G, Altissimi G, Simoncelli C, Ricci G. Sudden idiopathic
hearing loss. Case reports in the course of ten years. Acta Otorhinolaryngol Ital. 1998 Aug;18(4):218-27.
MECCANISMI DELL’ISCHEMIA J.-P. Sauvage, S. Puyraud, N. Khalifa. Sordità improvvise e fluttuanti . EMC – Otorinolaringoiatria 2005:1-16 [Article 20-183-A-10].
I fattori di rischio vascolari abitualmente incriminati a livello delle arterie di medio e grosso calibro perdono il loro valore per quanto riguarda il microcircolo cocleare. Tutti gli studi fatti alla ricerca di una tale correlazione con le sordità improvvise non hanno mai dimostrato nulla [ Mosnier et al., 1997 ] (fumo, alcol, ipertensione arteriosa [ IA ], diabete e iperlipidemie [ Ullrich et al., 1998 ]). Tuttavia, anche se non ci sono placche ateromasiche nei microvasi, microtrombi possono provenire da una placca situata a monte. Vi sono anche i casi di valvulopatie cardiache, del comune prolasso mitralico [ Pelisse et al 1988 ] (malattia di Barlow), e stati di ipertrombosi.
Essa è verosimile quando una sordità improvvisa compare nel corso di malattie che causano trombosi multiple (leucemia mieloide, disglobulinemia, anemia emolitica, drepanocitosi). È stato anche possibile dimostrare che l’incidenza della sordità improvvisa era più alta nei pazienti con precedenti di trombosi venosa profonda e portatori dell’allele 20210A. [ Mercier et al., 1999]
Uno studio mediante RMN avrebbe dimostrato un rallentamento del flusso sanguigno vertebrobasilare nel 21% dei pazienti di più di 50 anni con una sordità improvvisa. In tutti i casi si associavano vertigini. [ Yamasoba et al.,1993 ] Vari meccanismi possono essere portati in causa:
Alterazioni della viscosità ematica. Nei capillari e nelle micro arteriole di calibro inferiore a 100 μm, questa ha il suo effetto Fahraeus-Lindqvist; e cioè che, essendo la migrazione dei globuli assiale, questi sono separati dalla parete da uno strato plasmatico: così, la viscosità plasmatica assume un ruolo di primo piano. Nelle sordità improvvise, è stato messo in evidenza un aumento significativo della viscosità ematica e plasmatica. [ Ohinata et al., 1994 ] Tuttavia, tenuto conto del fatto che la viscosità dipende dalla concentrazione delle grandi molecole (fibrinogeno, α-2 macroglobulina e IgM), si sarebbe potuto ritenere che lo studio della concentrazione plasmatica in fibrinogeno fosse un fattore importante nell’eziologia e nel decorso delle sordità improvvise, cosa che non si è dimostrata vera.
Effetto «sludge». In caso di stasi inizia un’aggregazione intravascolare con formazione di pile d’emazie. Questo fenomeno dipende dalla viscosità del sangue, essa stessa funzione dell’ematocrito e della viscosità plasmatica. Questi fenomeni sono favoriti dall’ipotensione arteriosa [ Pirodda et al.,2001 ] e da una dieta ricca di lipidi. Questo stato è reversibile. Non avviene lo stesso per le piastrine, che formano aggregati irreversibili nel microcircolo. Nei pazienti con patologie cocleovestibolari sono state riscontrate iperaggregabilità piastrinica con ipercoagulabilità. Il ruolo della deformabilità delle emazie è altrettanto importante. Quando le emazie giungono in un capillare di diametro troppo sottile per il loro passaggio, si deformano. Nel corso di certe patologie ematologiche, la deformabilità del globulo rosso diminuisce e la circolazione capillare si blocca. È stata constatata una riduzione significativa della deformabilità delle emazie [ Hall et al., 1991 ] nelle sordità improvvise.
Ipotensione arteriosa sistemica. È stata oggetto di alcuni studi con monitoraggio della pressione arteriosa per 24 ore. In particolare, in soggetti d’età inferiore a 50 anni, senza fattori di rischio vascolari, si riscontrano crisi di ipotensione sistolica e diastolica diurne (70%) e notturne (87%) nei pazienti affetti da sordità improvvisa contro il 25% e 31% rispettivamente del gruppo controllo. Le sordità descritte sono soprattutto di tipo A. Generalmente questo viene interpretato come un fattore aggravante le cattive condizioni circolatorie nell’organo cocleare periferico.
Spasmi e sostanze vasoattive. È stata riscontrata nei periciti e nei miociti dei vasi della coclea la presenza di granulazioni citoplasmatiche ricche di istamina, serotonina, chinina e, soprattutto, prostaglandine. Queste sostanze sono vasoattive. In particolare, è stata messa in evidenza una prostaciclina in abbondanza a livello della parete esterna del canale cocleare. Essa potrebbe costituire uno dei mediatori della microcircolazione cocleare. [ Khonishi et al., 1998 ] In alcune condizioni patologiche compare anche l’endotelina (peptide fabbricato dall’endotelio con potere vasocostrittore importante). L’aspirina potrebbe così avere un ruolo tossico inibendo la sintesi di prostaciclina. In realtà, spasmo, trombi o emorragie sono intimamente correlati. La correlazione tra emicrania e sordità improvvisa potrebbe passare attraverso questo meccanismo.
Disturbi della regolazione del flusso cocleare. Le tecniche di studio nell’animale della microcircolazione cocleare sono complesse: microscopio intravitale, tecnica delle microsfere. Il laser-doppler è una tecnica che permette una valutazione istantanea globale e continua del flusso ematico cocleare. Lo studio dalla pressione in ossigeno della perilinfa e della clearance dell’idrogeno sono tecniche indirette. L’antipirina radioattiva permette uno studio autoradiografico della coclea. Gran parte del flusso ematico cocleare sembra seguire in maniera passiva le variazioni della pressione arteriosa. Alcuni parametri sono suscettibili di farlo variare: la caduta dell’ematocrito e l’inalazione di carbogeno (vedi. Trattamento). Esperimenti con fenilefrina suggeriscono un controllo della microcircolazione sul piano locale. Dei recettori α 1 e α 2– adrenergici sono stati evidenziati nella coclea, ma è ancora controverso il ruolo del sistema neurovegetativo nella regolazione della circolazione cocleare. Tuttavia è stato dimostrato che la stimolazione del simpatico cervicale o la simpatectomia potevano provocare nette variazioni del flusso ematico cocleare indipendenti dalla pressione arteriosa. [ Ren et al., 1993 ] Il nervo trigemino innerverebbe i vasi sanguigni cocleari. È un’altra possibilità di spiegare la maggiore frequenza di sordità improvvise negli emicranici. [ Vass et al 1998 ]
Essa potrebbe rivelarsi per l’importanza delle manifestazioni vestibolari associate e la sua comparsa in corso di trattamenti anticoagulanti, di emopatia o di aplasia midollare.
Anche se l’ipotesi vascolare è accettabile, essa rimane comunque impossibile da dimostrare nella clinica. Non può che essere una presunzione basata sul contesto. Esistono alcune osservazioni su soggetti deceduti che avevano presentato qualche giorno prima una sordità improvvisa e sui quali è stato possibile un esame istologico della rocca. Le lesioni sono le stesse di quelle riscontrate nell’animale dopo occlusione dell’arteria uditiva interna. [Schuknecht HF, Donovan ED 1986 ]
BIBLIOGRAFIA
Albers FW, Majoor MH, Van Der Gaag R Corneal autoimmunity in a patient with relapsing polychondritis. Eur Arch Otorhinolaryngol 1992 ; 249 : 296-299 [crossref]
Booth JB Sudden and fluctuant sensorineural hearing loss. A Kerr Scott-Brown’s otolaryngology. London: Butterworth-Heinemann: 1997; 1-82.
Hall SJ, McGuigan JA, Rocks MJ Red blood cell deformability in sudden sensorineural deafness: another aetiology. Clin Otolaryngol 1991 ; 16 : 3-7 [crossref]
Hultcrantz E Sudden deafness-A critical Evaluation of Pathogenesis and cure. Otorhinolaryngol Nova 1999 ; 9 : 178-189 [crossref]
Khonishi K, Yamane H, Igushi H Local substances regulating cochlear blood flow. Acta Otolaryngol [suppl] 1998 ; 553 : 40-46
Martin C, Mosnier I, Robier A, Bertholon P, Dubreuil C, Chobaut JC , e al. La pathologie vasculaire en ORL. Part du vasculaire dans les manifestations cochléo-vestibulaires. Rapport de la société française d’ORL 2000 : 237-258 [crossref]
Mazzoni A Internal auditory artery supply to the petrous bone. Ann Otol Rhinol Laryngol 1972 ; 81 : 13-21
Mercier E, Quere I, Chabert R, Lallemant JG, Daures JP, Berlan J , e al. The 20210A allele of the prothrombin gene is an independent risk factor for perception deafness in patients with venous thromboembolic antecedents [letter]. Blood 1999 ; 93 : 3150-3152
Mom T, Avan P, Bonfils P, Gilain L Vulnerability of the gerbil cochlea to sound exposure during reversible ischemia. Hear Res 1999 ; 136 : 65-74 [crossref]
Pelisse JM, Perles B, Jobert F, Charpy N, Fabiani F, Baril C À propos de 100 surdités brusques traitées en 10 ans : relations avec la maladie de Barlow. J Fr ORL 1988 ; 37 : 459-469
Pirodda A, Ferri GG, Modugno GC, Borghi C Systemic hypotension and the development of acute sensorineural haring loss in young healthy subjects. Arch Otolaryngol Head Neck Surg 2001 ; 127 : 1049-1052
Ren T, Laurikainen E, Quirk WS, Miller JM, Nuttal AL Effects on stellate ganglion stimulation on bilateral cochlear blood flow during increases in blood presure in human. Ann Otol Rhinol Laryngol 1993 ; 102 : 378-384
Schuknecht HF, Donovan ED The pathology of idiopathic sudden sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 1986 ; 243 : 1-15 [crossref]
Ueda T, Murai T, Nario K, Fujita N, Miyahara H, Matsunaga T Inner ear blood flow in the rat after unilateral occlusion in the vertebrobasilar system. Acta Otolaryngol [suppl] 1998 ; 533 : 36-39 [crossref]
Ullrich D, Aurbach G, Drobik C A prospective study of hyperlipidemia as a pathogenic factor in sudden hearing loss. Eur Arch Otorhinolaryngol 1992 ; 249 : 273-276 [crossref]
Vass Z, Shore SE, Nutall AL, Miller JM Direct evidence of trigeminal innervation of the cochlear blood vessels. Neuroscience 1998 ; 84 : 559-567 [crossref]
Yamasoba T, Kikuchi S, Yagi M, Higo R, O’Uchi T, Tokumaru A Sudden sensorineural hearing loss associated with slow blood flow of the vertebral system. Ann Otol Rhinol Laryngol 1993 ; 102 : 873-877
LE CAUSE VASCOLARI “P. R. Lazarini; A. C. Kfouri Camargo;Idiopathic sudden sensorineural hearing loss: etiopathogenic aspects Rev. Bras. Otorrinolaringol. vol.72 no.4 São Paulo July/Aug. 2006”
Il fatto che la coclea è principalmente fornita da un unico un’arteria, l’arteria labirintico, e questo è un’arteria terminale, rende l’orecchio interno molto incline ad alterazioni circolatorio Deppermann et al., 1998 25. Così, l’ipotesi vascolare è plausibile. Nonostante, se la SHL sono stati causati da una malattia vascolare, sarebbe ragionevole ritenere che i fattori di rischio e la fascia di età sia sarebbero simili, tuttavia, non sono.
Nelle cause vascolari, i problemi possono essere presenti sulla parete dei vasi sanguigni, come è il caso di arterite e spasmi, o intravascolari, come emboli gassosi, emboli di grasso, policitemia, iperviscosità (macrobulinemia di Waldenströn), crisi falciforme, e altri. Danno cocleare sarebbe secondaria di anossia o ipossia generata e potrebbe verificarsi a causa di tre diversi meccanismi Schuknecht HT & Donovan ED. 1986 14. Loro sono:
a) l’occlusione vasale totale e permanente;
b) l’occlusione temporanea totale e vasale;
c) Ipoafflusso di sangue Cocleare.
a)L’Occlusione totale e permanente di un vaso sanguigno causerebbe anossia, e quindi la necrosi delle membrane labirintico, fibrosi e ossificazione dell’orecchio interno. Ciò giustificare la perdita di udito, ma non il recupero di soglie tonali. Inoltre, rari sono i tempi in cui la proliferazione fibrosa e ossa sono stati trovati in qualche studio. Così, questa non è una teoria molto probabile che potrebbe giustificare la patogenesi ISHL.
b)La tolleranza della Cocleare all’ischemia è molto limitata e il potenziale d’azione comincia ad essere compromessa dopo 60 secondi di anossia Mattox DE & Simmons FB 1977 6. Quando c’è un blocco totale della circolazione, ma temporaneo, dopo 30 minuti le cellule ciliate, le cellule gangliari e il legamento spirale è già povero, inoltre, ci sono la perdita neuronale e piccola alterazione nella membrana tectoria. Quando il flusso di sangue torna alla normalità, i potenziali d’azione sono già irreversibilmente compromessi. Dopo un’ora di ostruzione vasale, non vi è più alcun recupero funzione cocleare, e dopo sei mesi vi è l’invasione del tessuto fibroso e osseo agli spazi dell’orecchio interno. Questo meccanismo spiega anche HL e anche, a seconda di quanto tempo l’occlusione vascolare dura, il recupero delle soglie tonali; tuttavia i reperti istopatologici sono ancora valutati poco frequentemente nelle ossa temporali dei casi SHL. Pertanto, anche questa è un’ipotesi improbabile per spiegare la fisiopatologia ISHL.
c)Basso flusso di sangue, il terzo meccanismo coinvolto, accade a causa di un iperviscosità, generando un calo di ossigenazione (insufficiente a mantenere il metabolismo cocleare) e la ipofunzione cocleare. Questa sarebbe la teoria più accettabile tra cause vascolari, dal momento che non solo giustifica HL, ma anche il possibile recupero audiologico e anche cocleari reperti istologici Vasama JP & Linthicum Jr FH. 2000 2, Loughan S. 2000 9, Schuknecht HT & Donovan ED., 2000 14.
Il flusso di sangue è inversamente proporzionale alla viscosità del sangue. La viscosità dipende l’ematocrito, la viscosità del siero, l’aggregazione dei globuli rossi e RCD. RCD è definita come una caratteristica fisico-chimico che consente al globulo rosso di passare attraverso capillari con diametri che variano tra 3 e 12μm, anche se in media il loro diametro è sempre sopra 7μm. Si ritiene che una riduzione differenziale può causare alcuni lesioni cocleari ed essere correlata alla SHL Hall et al., 1991 13.
L’ematocrito gioca un ruolo più importante nel flusso sanguigno dei grossi vasi, mentre RCD gioca un ruolo importante nella circolazione del flusso sanguigno, specialmente nei vasi più piccoli, e può essere influenzata in alcune situazioni, come le malattie coronariche, malattie del sangue e forma cambia cellulari, malattie renali, alterazioni post-operatorie e post AMI alterazioni, siero proteine alterazione, malattia vascolare periferica o diabete mellito. E ‘anche pensato che il fumo può avere influenza su RCD Hall et al., 1991 13.
Si ritiene che la riduzione differenziale può essere sia un fenomeno acuto o cronico, e sopporta grande incidenza nei casi di ISHL. Un schemia secondaria alla riduzione RCD potrebbe essere clinicamente confusa con una causa vascolare ostruttiva e spiegare la perdita di udito, e per di più un possibile recupero di soglie tonali in alcuni casi di ISHL. Da qui l’uso di farmaci che potrebbero migliorare la viscosità del sangue e RCD sarebbe giustificabile.
Ancora, sappiamo che una infezione acuta delle vie aeree superiori, insieme con una viremia, altera RCD e può portare ad una ischemia sul stria vascolare causa di un basso flusso di sangue e aumento shunt arterioso-venosa; sia l’ischemia e l’acidosi generati peggiorare riduzione RCD, provocando così un circolo vizioso. Pertanto, in caso di infezione acuta delle vie aeree superiori e riduce RCD è associato un viremia, la patogenesi ISHL può associare vascolare e fattori virali come origine ISHL Hall et al., 1991 13. Il virus può attaccare i globuli rossi e causa emoagglutinazione, causando uno stato di ipercoagulabilità, con un elevato consumo di protrombina in alcuni casi e, di conseguenza, occlusione vascolare temporanea 6. Un disturbo emotivo può anche innescare alterazioni nella viscosità del sangue e partecipare quindi nella eziopatogenesi ISHL . Fowler EP 1950 24.Quando tale alterazione si verifica in maniera transitoria, HL, vertigini e acufeni sono di breve durata, come in una crisi Ménière; se definitivo provoca HL irreversibile.
Fattori allergici, nonostante rare, possono anche causare cambiamenti nel microcircolo cocleare o, da fenomeni anafilattiche, e innescare un cambiamento nella permeabilità capillare. Pertanto, fisiopatologia in questi casi è davvero più legato ad una alterazione vascolare poi la allergico causa stessa Silvestre MN. 1999 3.
Il fatto della questione è che SHL sotto iperviscosità del sangue è molto controversa. Essendo che a causa di una riduzione del flusso di sangue nei capillari che alimentano vascularis stria e conseguentemente causare ischemia nell’orecchio interno, o una riduzione della capacità di trasporto di ossigeno a causa di un lento flusso di sangue e la conseguente generazione di micro trombi e lesioni al endotelio cocleare, trombosi al sistema venoso dell’orecchio interno; o qualsiasi altra ipotesi, è evidente che non c’è modo assoluto per dimostrare Deppermann et al., 1998 25. È noto, tuttavia, che tutte le manifestazioni cliniche della sindrome da iperviscosità rispondono alla plasmaferesi.
L’emorragia orecchio interno potrebbe anche essere una spiegazione importante per ISHL. Secondaria l’uso di anticoagulanti o malattie che causano bassi livelli di piastrine, come la leucemia e l’anemia aplastica, traumi, la meningite, emorragia subaracnoidea e adenocarcinoma metastasi, questa emorragia sarebbe allora causare un aumento della pressione improvviso della endolinfa o alterazioni perilymph, biochimiche e osmolarità nei fluidi intracochlear, intracochlear disturbi della conduzione e ischemia della regione periferica accanto al sito sanguinamento. Alterazioni istologiche trovate nel cocleare di questi pazienti trombocito penici mostrare e dimostrare come sanguinamento nelle peri e spazi endolinfatico, oltre infiltrato cellulare. Nonostante, considerando che l’emorragia spazio perilinfatico è responsabile SHL, i meccanismi non sono così chiare come sembrano, dal momento che molti argomenti li contraddicono. Non abbiamo notato alcuna perdita di udito sangue indotta nel perilinfa durante stapedectomia; nessuno studio istologico mostra i danni alle strutture cocleari causati dalla presenza di sangue nello spazio perilinfatico dopo stapedectomia; e anche dopo l’infusione sperimentale di sangue nel perilymph suini; nessun potenziale ampiezza o alterazione della soglia è stato notato sul elettrococleogramma. Pertanto, nemmeno un’emorragia insidiosa nello spazio perilinfatico potrebbe causare SHL: determinando così un enorme situazione di stallo. Nonostante una non molto chiaro fisiopatologia, sembra che ci sia un rapporto intimo tra le patologie di cui sopra e la loss Ogawa K & Kanzaki J 1994 26 dell’udienza, avendo visto che in molti test di risonanza magnetica è molto presente tale accertamento (sanguinamento orecchio interno).
Eppure, tra le cause vascolari, è noto che le diete occidentali offrono un maggiore rischio di ISHL rispetto alla dieta giapponese, dato che il nostro è più caricato con grassi saturi. Dieta occidentale provoca maggiore aggregazione piastrinica e l’attività VII fattore, aumentando coagulabilità del sangue e il rischio di ostruzione vascolare. Ora, la dieta orientale, ricca di oli vegetali, è più importante antitrombotica e le proprietà antiaterosclerotici che prevengono malattie vascolari Nakamura et al.,200127.
Così, una storia di malattia vascolare o alterazioni delle piastrine o dei globuli rossi sono gli unici risultati clinici che ci potrebbero portare a inferire una causa vascolare per ISHL; Istopatologico, dovremmo trovare la proliferazione fibrosa e ossea negli spazi cocleari e canali semicircolari, o per lo meno, l’emorragia negli spazi cocleari, dimostrano non solo studi di immagine, ma anche da post mortem scansioni tomografiche Schuknecht HT & Donovan ED 1986 14.Potrebbero esistere Altri risultati istologici, quali Corti organo, stria vascularis, spirale legamento e neuroni cocleari degenerazione: è probabile che tali differenze sono dovute al grado di coinvolgimento vascolare o differenze nel meccanismo patogenetico . Yoon et al.,1990 28.
REFERENCES
1. Caldas Neto & Caldas Neto S. Surdez súbita. In: Lopes Filho OL & Campos CAH. Tratado de otorrinolaringologia. São Paulo: Roca; 1994. p. 869-80. [ Links ]
2. Vasama JP & Linthicum Jr FH. Idiopathic sudden sensório-neural hearing loss: temporal bone histopathologic study. Ann Otol Rhinol Laryngol 2000;109:527-32. [ Links ]
3. Silvestre MN. Surdez súbita. Recife; 1999. (Monografia – CEFAC – Centro de especialização em Fonoaudiologia Clínica). [ Links ]
4. Byl FM. Sudden hearing loss: eight years’ experience and suggested prognostic table. Laryngoscope 1984 May;94:647-61. [Links ]
5. Hallberg OE. Sudden deafness of obscure origin. Laryngoscope 1956;66(10):1237-67. [ Links ]
6. Mattox DE & Simmons FB. Natural history of sudden sensorioneural hearing loss. Ann Otol 1977;86:463-80. [ Links ]
7. Vakkalanka S, Ey E, Goldenberg RA. Inner ear hemorrhage and sudden sensorioneural hearing loss. Am J Otol 2000;21:764-5. [ Links ]
8. Stokroos RJ, AlbersFWJ, Tenvergert EM. Antiviral treatment of idiopathic sudden sensorioneural hearing loss: a prospective, randomized, double – blind clinical trial. Acta Otolaryngol (Stockh) 1998b;118:488-95. [ Links ]
9. Loughan S. Management of sudden sensorioneural hearing loss: a consultant survey. J Olaryngol Otol 2000 June;114:837-9. [ Links ]
10. Stokroos RJ, AlbersFWJ, Schirm J. Therapy of Idiopathic Sudden Sensorioneural Hearing Loss: Antiviral treatment of experimental herpes simplex virus infection of the inner ear. Ann Otol Rhinol Laryngol 1999;108:423-8. [ Links ]
11. Fukuda S, Furuta Y, Takasu T, Suzuki S, Inuyama Y, Nagashima K. The significance of herpes viral latency in the spiral ganglia. Acta Otolaryngol Suppl (Stockh) 1994;514:108-10. [ Links ]
12. Pitkaranta A & Julkunen I. Sudden deafness: lack of evidence for systemic viral infection. Otolaryngol Head Neck Surg 1998;118(3 Pt 1):397-9. [ Links ]
13. Hall SJ, McGuigan JA, Rocks MJ. Red blood cell deformability in sudden sensorioneural deafness: another aetiology? Clin Otolaryngol 1991 June;16:3-7. [ Links ]
14. Schuknecht HT & Donovan ED. The pathology of idiopathic sudden sensorioneural hearing loss. Arch Otorhinolaryngol 1986;243:1- 5. [ Links ]
15. Wilson WR. The relationship of the herpesvirus family to sudden hearing loss: a prospective clinical study and literature review. Laryngoscope 1986 Aug;96:870-7. [ Links ]
16. Wackym PA. Molecular temporal bone pathology: II. Ramsay Hunt syndrome (herpes zoster oticus). Laryngoscope 1997;107:1165-75. [ Links ]
17. Cadoni G, Marinelli L, De Santis A, Romito A, Manna R, Ottaviani F. Sudden hearing loss in a patient hepatitis C virus (HCV) positive on therapy with alpha-interferon: a possible autoimmune-microvascular pathogenesis. J Laryngol Otol 1998; 112:962-3. [ Links ]
18. Cole RR & Jahrsdoerfer RA. Sudden hearing loss: an update. Am J Otol 1988;9:211-5. [ Links ]
19. Yoshida Y, Yamauchi S, Shinkawa A, Horiuchi M, Sakai M. Immunological and virological study of sudden deafness. Auris Nasus Larynx 1996;23:63-8. [ Links ]
20. Murai K, Tsuiki T, Shishido K, Hori A. Clinical study of sudden deafness with special reference to onset. Acta Otolaryngol (Stockh) 1988; Suppl456:15-20. [ Links ]
21. Berrocal JRG, Camacho RR, Portero F, Vargas JA. Role of viral and Mycoplasma pneumoniae infection in idiopathic sudden sensorioneural hearing loss. Acta Otolaryngol 2000; 120:835-9. [ Links ]
22. Fitzgerald DC & Mark AS. Sudden hearing loss: frequency of abnormal findings on contrast-enhanced MR studies. AJNR Am J Neuroradiol 1998 Sept;19:1433-36. [ Links ]
23. Oliveira WD, Pinto JÁ, Becker CG, Becker HMG, Guimaraes, RES, Rezende AL. Hipoacusia neurosensorial súbita imunomediada: relato de caso. Rev Bras Otorrinolaringol 2005;71(3):41-5. [ Links ]
24. Fowler EP. Sudden deafness. Ann Otol Rhinol Laryngol 1950;59:980-7. [ Links ]
25. Deppermann MB, Nadaf LC, Caron C, Contini JE, Amarante LF. Surdez súbita bilateral por macroglobulinemia de Waldeströn. Rev Bras Otorrinolaringol 1998;64 (2 Pt 1):165-8. [ Links ]
26. Ogawa K & Kanzaki J. Aplastic anemia and sudden sensorioneural hearing loss. Acta Otolaryngol Suppl (Stockh) 1994;(Suppl 514):85-8. [ Links ]
27. Nakamura M, Whitlock G, Aoki N, Nakashima T, Hoshino T, Yokoyama T et al. Japanese and western diet and risk of idiopathic sudden deafness: a case-control study using pooled controls. Int J Epidemiol 2001; 30:608-15. [ Links ]
28. Yoon TH, Paparella MM, Schachern PA, Alleva M. Histopathology of sudden hearing loss. Laryngoscope 1990;100(7):707-15. [ Links ]
29. Bittar RSM, Thomé DC, Nascimento EV, Sanchez TG. Doenças auto-imunes da orelha interna: revisão da literatura. Arq. Otorrinolaringol. 1998; 2(3): 92-9. [ Links ]
30. Alvarenga EHL, Cruz OLM, Grellet M, Colafëmina JF. Disacusia sensório-neural auto-imune: avaliação auditiva em pacientes portadores de doença auto-imune. Rev Bras Otorrinolaringol 1999;65:50-7. [ Links ]
31. McCabe BF. Autoimmune sensorioneural hearing loss. Ann Otol 1979;88:585-9. [ Links ]
32. Yanagita N, Koide J, Yokoi H. Morphological changes in the cochlear aqueduct following herpes simplex virus inoculation into the subarachnoid space. Acta Otolaryngol Suppl (Stockh) 1988;Suppl456:106-10. [ Links ]
33. Al Muhaimeed H & Zakzouk SM. Hearing loss and herpes simplex. J Trop Pediatr 1997 Feb;43:20-4. [ Links ]
34. Fuse T, Inamura H, Nakamura T, SuzukiI T, Aoyagi M. Bilateral hearing loss due to viral infection. ORL J Otorhinolaryngol Relat Spec 1996 Sept;58:175-7. [ Links ]
35. Leitner C. Does virus diagnosis open new ways for the classification and treatment of sudden deafness, unilateral vestibular loss and idiopathic facial paralysis? HNO 1986;34:376-8. [ Links ]
36. Yanagita N & Murashi K. A comparative study of mumps deafness and idiopathic profound sudden deafness. Arch Otorhinolaryngol 1986;243:197-9. [ Links ]
37. Stokroos RJ, AlbersFWJ, Schirm J. The etiology of idiopathic sudden sensorioneural hearing loss. Experimental herpes simplex virus infection of the inner ear. Am J Otol 1998a;19:447-52. [ Links ]
38. Lindsay JR & Zuidema JJ. Inner ear deafness of sudden onset. Laryngoscope 1950;60:238-63. [ Links ]
39. Toubi E, Ben-David J, Kessel A, Podoshin L, Golan TD. Auto-imune aberration in sudden sensorioneural hearing loss: association with anti-cardiolipin antibodies. Lupus 1997;6:540-42. [ Links ]
Meccanismi Patogenetici Dei Disordini Vascolari Orecchio Interno

L’eziologia vascolare è stata dedotta da:
1. Rilevazione anamnestica di almeno tre dei fattori di rischio cardiovascolari selezionati: EVENTI VASCOLARI CEREBRALI: (TIA-Ictus/Insufficienza Vertebro-Basilare/patologia carotidea); NON CEREBRALI: cardiopatia ischemica(Angina-IMA/fibrillazione atriale/arteriopatia periferica nelle sue espressioni di claudicatio intermittens e di dolore a riposo), ASSENZA DI EVENTI VASCOLARI: Dislipidemia- Diabete Mellito- Ipertensione Arteriosa, arteriopatia periferica, familiarità per patologia vascolare, Ipercoagulazione Fattori Coagulativi:[Fattore V Leiden Protrombina G20210A – Iperfibrinogemia] Anticoagulanti fisiologici :[AT III-Proteina C/S], fumo, consumo di alcool, obesità, fibrinogenemia >350 mg/dL, ipertrigliceridemia (>180 mg/dL) ed ipercolesterolemia (>220 mg/dL).
I fattori di rischio più rappresentati sono stati: ipertensione arteriosa (71,7%), ipercolesterolemia (64,1%), patologia carotidea (45,7%) e familiarità per malattie cardiovascolari (59,7%).
2 Chiara positività per patologia vascolare di almeno uno dei seguenti esami: eco Doppler dei vasi sovra-aortici, TC o RM cerebrale.
Fattori che possono aumentare la viscosità del sangue
· Aumento della viscosità del plasma Aumento della viscosità del plasma
· Aumento del numero dei globuli rossi Aumento del numero dei globuli rossi
· Aumentata aggregabilità dei globuli rossi
· Aumentata aggregabilità dei globuli rossi
· Diminuzione della deformabilità dei globuli rossi Diminuzione della deformabilità dei globuli rossi
Iperviscosità plasmatiche
La viscosità plasmatica varia al variare della concentrazione di alcune proteine, in particolare del fibrinogeno in relazione al suo elevato peso molecolare
· Iperfibrinogenemie
· Waldeström
· Collagenopatie
· Crioglobulinemie
· Mieloma multiplo
Mieloma multiplo Nel mieloma multiplo la viscosità è normalmente più alta rispetto a quella dei controlli normali, ma è spesso compensata dallanemia (ematocrito ridotto)
Iperviscosità da aumentata concentrazione dei globuli rossi
Policitemia vera Poliglobulie secondarie
In tutti i pazienti con policitemia vera, con o senza terapia citoriduttiva è consigliabile mantenere il valore dell Hct al di sotto del 45% per ridurre al minimo il rischio tromboembolico In tutti i pazienti con policitemia vera, con o senza terapia citoriduttiva è consigliabile mantenere il valore dell Hct al di sotto del 45% per ridurre al minimo il rischio tromboembolico
Iperviscosità da anormalità cellulari
· Sferocitosi
· Anemie falciformi
· Leucemie
· Emoglobinopatie
Iperviscosità secondarie
· Diabete
· Neoplasie
· Fumo
· Disidratazione
· Disidratazione
· Shock ipovolemico
· Ipertensione
· Iperdislipidemie
Cause Autoimmuni e Vascolari dei Disordini dell’Orecchio Interno
|
Autoimmune |
Vascolare |
Neurologico neoplastica |
Trauma o Tossina |
Infezioni e virale |
|
Bypass cardiopolmonare |
Meningite criptococcica |
|||
|
La deformabilità dei globuli rossi |
Sordità controlaterale dopo la chirurgia neuroma acustico |
Interno commozione cerebrale orecchio |
Citomegalovirus |
|
|
Lupus |
Falciforme |
Focal ischemia pontino |
Malattia da decompressione dell’orecchio interno |
Herpes simplex-I |
|
Malattia dei piccoli vasi |
Leucemia |
Chirurgia otologica |
HIV |
|
|
Poliartrite nodosa |
Malattia vascolare associata mitocondriopatia |
Carcinosi meningea |
Febbre di Lassa |
|
|
Policondrite |
Fistola Perilinfatica |
Meningite meningococcica |
||
|
Colite ulcerosa |
Discrasie ematiche |
Sclerosi multipla |
Parotite |
|
|
Granulomatosi di Wegeners |
Mieloma |
Perdita CSF, come ad esempio causati da puntura lombare |
Rubeola, rosolia, la sifilide, toxoplasmosi |
Patologie associate ad alterazioni del flusso ematico
Oltre all’ipoacusia improvvisa, di cui abbiamo già parlato, sono molte le patologie dell’orecchio interno che riconoscono una base vascolare; qui di seguito vengono analizzate le principali con alcune considerazioni generali(11).
Noise-Induced Hearing Loss. Tra i diversi meccanismi fisiopatologici che sostengono l’ipoacusia transitoria o permanente da esposizione al rumore, bisogna sicuramente includere la riduzione del flusso sanguigno cocleare; diversi studi hanno dimostrato che l’esposizione al rumore comporta una vasocostrizione dei capillari della membrana basilare, del legamento spirale e della stria vascolare. È stato inoltre accertato un aumento della pressione arteriosa sia nell’uomo sia nell’animale, accompagnata da alterazioni del metabolismo del magnesio. Il magnesio sembra volgere un ruolo protettivo nei confronti del rumore. Un’esposizione prolungata a suoni non confortevoli può aumentare notevolmente la produzione di radicali liberi, contribuendo alla patogenesi dell’ipoacusia da rumore. Da queste considerazioni iniziali, si è ipotizzato che la tipica lesione nel profilo audiometrico a 4-6 kHz sia attribuibile all’area di massima sollecitazione della membrana basilare, con conseguente massima riduzione del flusso locale.
Idrope endolinfatica. Sebbene molti autori non abbiano riscontrato differenze significative nel flusso sanguigno di coclee con idrope rispetto a coclee normali, è stata accertata un’alterazione dell’autoregolazione locale : l’idrope endolinfatica riduce il flusso sanguigno cocleare.
Presbiacusia. Che all’invecchiamento possano associarsi alterazioni del flusso locale e sistemico è risaputo; tuttavia non sono chiare le relazioni temporali e causali che portano all’ipoacusia e alla degenerazione delle strutture vascolari.
Acquedotto vestibolare allargato. Il rapporto tra perfusione cocleare e pressione dei fluidi cocleari è identico a quello esistente tra perfusione cerebrale e liquido cerebrospinale, per cui all’aumentare della pressione dei fluidi si ha una riduzione del flusso sanguigno. L’acquedotto vestibolare o cocleare allargati rappresentano condizioni patologiche con aumento della pressione dei fluidi dell’orecchio interno; la conseguenza di ciò è una riduzione del flusso sanguigno cocleare. D’altro canto, anche in situazioni opposte si può avere ipoacusia: una drastica e rapida riduzione della pressione dei fluidi può comportare danno della funzione uditiva anche in presenza di un adeguato flusso arterioso.
Emopatie. Come già accennato, tutte quelle condizioni che aumentano la viscosità ematica possono comportare un danno da ipoperfusione cocleare: leucemia, leucocitosi, policitemia, anemia falciforme, crioglobulinemia e macroglobulinemia sono tutte condizioni che possono alterare il flusso sanguigno cocleare e l’ipoacusia può essere il primo sintomo della malattia. Queste patologie inoltre possono portare ad una fibrosi degli spazi perilinfatici e causare episodi di microembolismo.
Disordini dei vasi sanguigni. Dopo quanto riportato finora, non ci si stupirà del fatto che tutti i disordini dei vasi, dalle vasculiti autoimmuni alle sindromi congenite, possono avere ripercussioni importanti sull’organo dell’udito. Anche il
diabete com’è noto, soprattutto negli stadi più avanzati, può portare ad un danno notevole del microcircolo di tutti i distretti anche di quello cocleare, con perdita della capacità di autoregolazione ed ispessimento delle pareti dei vasi(12).
Tra le altre patologie che riconoscono una base vascolare nell’insorgenza dell’ipoacusia vale la pena ricordare la Susac Syndrome, la Norrie Syndrome e l’arterite di Takayasu.
Occlusione del drenaggio venoso. Sebbene sia lecito pensare che un’occlusione del drenaggio venoso possa influire negativamente sul flusso sanguigno coclea-re, studi condotti sugli animali non hanno dimostrato una significativa associazione tra occlusione delle vene dell’orecchio interno ed insorgenza di ipoacusia.
Bibliografia
11) Mudry A, Tange RA. The vascularization of the human cochlea: its his‑
torical background. Acta Otolaryngol Suppl. 2009;(561):3-16.
12) Delas B, Dehesdin D. Anatomia dell’orecchio esterno. EMC (Elsevier
Masson SAS), Otorinolaringoiatria, 20-010-A-10, 2008.
13) Thomassin JM, Dessi P, Danvin JB, Forman C. Anatomia dell’orecchio
medio. EMC (Elsevier Masson SAS), Otorinolaringoiatria, 20-015-A-10, 2008.
14) Sauvage JP, Stéphane Puyraud S, Roche O, Rahman A. Anatomia
dell’orecchio interno. EMC (Elsevier Masson SAS), Otorinolaringoiatria, 20-020-A-10, 2002.
15) Ferrari Lelli G. Comportamento dell’arteria uditiva interna e dei suoi
rami labirintici nell’uomo. Anatomy and Embryology, 1939; 110, 1:48- 80.
16) Mevio E, Richichi M. Le ipoacusie improvvise. Quaderni Monografici di Aggiornamento A.O.O.I. 2009.
17) Mannini L, Paniccia R, Cecchi E, Alessandrello Liotta A, Leprini E, Berloco
P, Pagnini P, Abbate R, Franco Gensini G, Prisco D. Reduced erythrocyte deformability and hypercoagulability in idiopathic sudden sensorineural hearing loss. Clin Hemorheol Microcirc. 2005;33(1):47-55.
18) Arima T, Uemura T, Yamamoto T. Structural features of the basal lami‑
na in Reissner’s membrane of the guinea pig. Acta Otolaryngol. 1985 Sep-Oct;100(3-4):194-200.
19) Quick CA, Fish A, Brown C. The relationship between cochlea and kid‑
ney. Laryngoscope. 1973 Sep;83(9):1469-82.
20) Dai M, Shi X. Fibro-vascular coupling in the control of cochlear blood flow. PLoS One. 2011;6(6):e20652.
19) Nakashima T, Naganawa S, Sone M, Tominaga M, Hayashi H, Yamamoto
H, Liu X, Nuttall AL. Disorders of cochlear blood flow. Brain Res Brain Res Rev. 2003 Sep;43(1):17-28.
20) Kariya S, Cureoglu S, Fukushima H, Morita N, Baylan MY, Maeda Y,
Nishizaki K, Paparella MM. Comparing the cochlear spiral modiolar artery in type-1 and type-2 diabetes mellitus: a human temporal bone study. Acta Med Okayama. 2010 Dec;64(6):375-83.
21) Weiss G, Werner-Felmayer G, Werner ER, Grünewald K, Wachter H,
Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. 1994 September 1; 180(3): 969–976.
22) Wolff NA, Liu W, Fenton RA, Lee WK, Thévenod F, Smith CP. Ferroportin
1 is expressed basolaterally in rat kidney proximal tubule cells and iron excess increases its membrane trafficking. J Cell Mol Med. 2011 Feb;15(2):209-19.
23) Dai M, Yang Y, Omelchenko I, Nuttall AL, Kachelmeier A, Xiu R, Shi X.
Bone Marrow Cell Recruitment Mediated by Inducible Nitric Oxide Synthase/Stromal Cell-Derived Factor-1α Signaling Repairs the Acoustically Damaged Cochlear Blood-Labyrinth Barrier. Am J Pathol. 2010 Dec;177(6):3089-99.
24) Chen CY, Emmerling O, Ilgner J, Westhofen M. Idiopathic sudden sen‑
sorineural hearing loss in children. Int J Pediatr Otorhinolaryngol 2005;69:817-821.
25) Cole RR, Jahrsdoerfer R. Sudden hearing loss. Am J Otol 1988;9:211-
215.
26) Nakashima T, Tanabe T, Yanagita N, Wakai K, Ohno Y. Risk factors for
sudden deafness: a case control study. Aurus Nasus Larynx 1997;24:265-270.
31) Zadeh M H, Storper I.S, Spitzer JB. Diagnosis and treatment of sudden‑
onset sensorineural hearing loss: A study of 51 patients. Otolaryngol Head Neck Surg 2003;128:93-98.
32) Kroenenberg J. Vasoactive therapy versus plasebo in the treatment of
sudden hearing loss: A double-blind clinical study. Laryngoscope 1992;102.
33) Matteson EL Use of methotrexate for autoimmune hearing loss. Ann
Otol Rhinol Laryngol 2000;109:710-714.
34) Chen CY, Halpin C, Rauch SD. Oral steroid treatment of sudden senso‑
rineural hearing loss: a ten year retrospective analysis. Otol Neurotol 2003;24:728-733.
35) Weinaug P. Spontaneous remission in sudden deafness. HNO 1984;32:346-351.
36) Mattox D E, Simmons FB. Natural history of sudden sensorineural
hearing loss. Ann Otol Rhinol Laryngol 1977;86:463-480.
37) SnowJB Jr. Sudden deafness. In: Paparella MM, Shumrick DA, eds. Oto‑
laryngology. Philadelphia: W.B. Saunders, 1980:1757-1766.
38) Mattox DE, Lyles CA. Idiopathic sudden sensorineural hearing loss. Am
J Otol 1989; 10:242-247.
39) Psifidis AD, Psillas GK, Daniilidis JCh. Sudden sensorineural hearing loss:
Long-term follow-up results. Otolaryngol Head Neck Surg 2006;134:809-815.
40) Shaia FT, Sheehy JL. Sudden sensorineural hearing impairment: A re‑
port of 1220 cases. Laryngoscope 1976;86:389-398.
41) Laird N, Wilson WR. Predicting recovery from idiopathic sudden hear‑
ing loss. Am J Otolaryngol 1983;4:161-165.
42) Byl FM. Sudden hearing loss: eight years’ experience and suggested
prognostic table. Laryngoscope 1984;94:647-661.
40) Fetterman B, Saunders J, Luxford W. Prognosis and treatment of sud‑
den hearing loss. Am J Otol 1996;17:529-536.
41) Huy PT, Sauvaget E. Idiopathic sudden sensorineural hearing loss is not an otologic emergency. Otol Neurootol 2005;26:896-902.
42) Aimoni C, Bianchini C, Borin M, Ciorba A, Fellin R, Martini A, Scanelli G,
Volpato S. Diabetes, Cardiovascular Risk Factors and Idiopathic Sudden Sensorineural Hearing Loss: A Case-Control Study. Audiol Neurotol 2010;15:111–115
43) Gemmati D, Federici F, Catozzi L, Gianesini S, Tacconi G, Scapoli GL,
Zamboni P. DNA-array of gene variants in venous leg ulcers: detection of prognostic indicators. J Vasc Surg. 2009 Dec;50(6):1444-51.
44) Zamboni P, Gemmati D. Clinical implications of gene polymorphisms in
venous leg ulcer: a model in tissue injury and reparative process. Thromb Haemost. 2007 Jul;98(1):131-7. Review.
45) Gemmati D, Tognazzo S, Catozzi L, Federici F, De Palma M, Gianesini S,
Scapoli GL, De Mattei M, Liboni A, Zamboni P. Influence of gene polymorphisms in ulcer healing process after superficial venous surgery. J Vasc Surg. 2006 Sep;44(3):554-62.
46) Zamboni P, Izzo M, Tognazzo S, Carandina S, De Palma M, Catozzi L,
Caggiati A, Scapoli G, Gemmati D. The overlapping of local iron overload and HFE mutation in venous leg ulcer pathogenesis. Free Radic Biol Med. 2006 May 15;40(10):1869-73.
47) Zamboni P, Tognazzo S, Izzo M, Pancaldi F, Scapoli GL, Liboni A, Gem‑
mati D. Hemochromatosis C282Y gene mutation increases the risk of venous leg ulceration. J Vasc Surg. 2005 Aug;42(2):309-14.
48) Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews
NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005 Mar;1(3):191-200.
PATOLOGIE DEI VASI ADIACENTI: Nadir Yıldırım Hearing Impairment in Vascular Disorders Van Tıp Dergisi: 19 (3): 149-157, 2012
Disturbi vascolari congeniti o idiopatici:
a. Una Carotide Aberrante all’interno dell’orecchio come risultato di un ipotimpano deiscente può causare una perdita uditiva mista di gravità variabile, tuttavia è una condizione molto rara (22, 23, 24).
b. Loop Arterioso dell AICA comprimono a volte l’VIII° nervo, causa una sintomatologia tipica dell’Angolo pontine cerebellare (CPA) i risultati di massa con i segni e sintomi di sordità neurosensoriale (SNHL) di provenienza retro-cocleare (25, 26).L’acufene pulsane è anche riferito come una delle caratteristiche di questa variante anatomica (27).
c. l’ Ectasia Vertebrale e dell’arteria Basilare è stato dimostrata che imita i tumori CPA, la malattia di Ménière e le altre condizioni con sintomi dell’orecchio interno periferiche o centrali, comprimendo il tronco encefalico e l’VIII °nervo (26, 28).
d. Aneurisma dell’ AICA può anche causare perdita dell’udito improvvisa, anche se è estremamente raro, soprattutto proviene da loop di tutto il canale uditivo interno (IAM). meno di 50 casi sono stati segnalati finora. I segni ed i sintomi sono indistinguibili dai tumori CPA (29, 30).
e. Fossa Giugulare Alta di solito è osservata sul lato destro, invade il labirinto, ed interferisce sull’acquedotto cocleare, acquedotto vestibolare ed erode il canale semicircolare posteriore. Essa provoca in genere SNHL per le frequenze basse e occasionale ipoacusia trasmissiva (31, 32).
f. Fossa Giugulare Notevolmente ampliato si trova anche ad un livello superiore al normale ed è accompagnato da un seno sigmoideo gigante e talvolta con un diverticolo. Sintomatologia e l’etio-patogenesi è molto simile a quelli delle fossa giugulare alta . Inoltre, fossa giugulare gigante può influenzare la circolazione cocleare causando flusso turbolento e un ritorno venoso diminuito (31).
g. Malformazione cavernosa del interna canale acustico (33).
h. Dissezione dell’arteria vertebrale (34, 35,36).
i. Carotide Anomala (37).
j. Anomalie arteriose dell’orecchio medio associata con anchilosi della staffa (38).
K. Aneurisma derivanti dalla porzione petrosa della carotide interna (39).
I. Infarto dell’arteria uditivo interna :nell’ infarto isolato dell’arteria uditiva interna sono anche stati riportati reperti ‘istopatologici che correlano con la degenerazione del nervo vestibulo-cocleare (40).
m. Sindrome di Susac è un disturbo idiopatico caratterizzata dalla triade encefalopatia, perdita dell’udito fluttuante, [ sordità improvvisa ,Acufeni intensi, Vertigini e altri disturbi dell’equilibrio, Recruitment, difficoltà di discernimento delle parole, riduzione della soglia di tolleranza del rumore]. perdita della vista e conseguente microangiopatia cerebrale, cocleare e retinica ipoacusia neuro-sensoriale (41).
Tumori vascolari:
a. Emangio-endotelioma dell’osso temporale(42).
b. Emangioma cavernoso del IAC (43).
c. I tumori glomo-giugulari (paragangliomi) causano una ipoacusia trasmissiva nelle fasi iniziali e può invadere labirinto, causando SNHL (44).
d. Lesioni non paragangliomatose del forame giugulare(45).
e. Angiosarcoma dell’osso temporale (46).
g. Schwannoma del forame Giugulare (47).
vasculite sistemica:
Qualsiasi malattia vascolare sistemica che coinvolge piccole arterie ed arteriole può influenzare facilmente vasi cocleari. Essi comprendono una ampio spettro delle malattie che sono caratterizzata da vasi sanguigni disseminata infiammazione in diversi organi e sistemi, per lo più di vari meccanismi immunopatologici. La Vasculite sistemica provoca di solito improvvisa o progressiva SNHL o uditiva mista perdita predisponendo diverse forme di otite supporto, come pure (63). Una volta che influenzano l’orecchio interno,il danno è di solito grave e irreversibile.
Insufficienze arteriolare sistemica di lunga data tendono a causare la perdita dell’udito a bassa frequenza, presumibilmente a causa risultante atrofia striale, considerando che le malattie cardiovascolari di cui sopra in genere causare o contribuire ad alta frequenza perdita da perdita di cellule ciliate udito.
a. Il Lupus Eritematoso Sistemico (LES): in genere provoca grave vasculite nel giro apicale della coclea. Le cellule ciliate esterne sono perse per necrosi ischemica e microinfarti. Manifestazione clinica tipico è SNHL a rapida progressione (63, 65).
b. Granulomatosi di Wegener: provoca comunemente otite media con effusione (35-47%), a volte con cambiamenti granulomatose cronici dell’orecchio medio (64).Può anche causare danni cocleare e danni all VIII° nervo influenzando direttamente le loro arteriole attraverso una vasculite necrotizzante (66).
poliarterite nodosa (PAN) . tende a provocare alterazioni più gravi dell’orecchio interno, che a volte porta a infarti e cambiamenti fibrotici e la forma meno grave di OME di granulomatosi di Wegener (65).
d. Sindrome di Cogan: vasculite attiva e fibrosi delle arterie medie e piccole sono tipici di questo immuno-mediata malattia idiopatica. Il sistema Audio-vestibolare è spesso coinvolto con vertigini associate, acufeni e perdita dell’udito (67, 68).
e. Tromboangite Obliterante possono causare improvvise perdita dell’udito (52).
f. Sclerosi Sistemica (Sclerodermia): In una studio particolare, il 77% dei casi con sclerosi sistemica mostra livelli acustici anormalmente bassi (69).
References
1. Yamasoba T, Kikuchi S, O`uchi T, Higo R, Tokumaru A. Sudden sensorineural hearing loss associated with slow blood flow of vertebrobasilar system. Ann Otol Rhinol Laryngol 1993; 102: 873-877.
2. Lownie SP, Parnes LS. Isolated vestibulocochlear dysfunction of central or peripheral origin. Laryngoscope 1991; 101:1339-1342.
3. Donaldson JA. Duckert LG. Anatomy of the ear Hearing in Vascular Disorders Derleme Van Tıp Dergisi, Cilt:19, Sayı:3, Temmuz/2012 155 in “Otolaryngology”. Eds: Paparella MM, Shumrick DA, Gluckman JL, Meyerhoff WL, third ed. WB. Saunders; Philadelphia, PA 1991:23-58.
4. Sidman JD, Prazma J, Pulver S, Pillsbury HC. Cochlea and heart as end-organs in small vessel disease. Ann Otol Rhinol Laryngol 1988; 97:9- 13.
5. Hawkins JE. The role of vasoconstriction in noise induced hearing loss. Ann Otol Rhinol Laryngol 1971; 80:903-913.
6. Hildesheimer M, Bloch F, Muchnik C, Rubinstein M. Blood viscosity and sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 1990; 116: 820-823.
7. Wangemann P, Wonneberger K. Neurogenic regulation of cochlear blood flow occurs along the basilar artery, the anterior inferior cerebellar artery and at branch points of the spiral modiolar artery. Hear Res 2005; 209:91-96.
8. Brown JN, Nuttall AL. Autoregulation of cochlear blood flow in guinea pigs. Am J Physiol 1994 ; 266:458-467.
9. Franz P, Helmreich M, Stach M, Franz-Italon C, Böck P. Distribution of actin and myosin in the cochlear microvascular bed. Acta Otolaryngol 2004; 124:481-485.
10. Bachor E, Selig YK, Jahnke K, Rettinger G, Karmody CS. Vascular variations of the inner ear. Acta Otolaryngol 2001; 121:35-41.
11. Axelsson A and Dengerink H. The effects of noise on histological measures of the cochlear vasculature and red blood cells: A review. Hearing Research 1987; 31:183-192.
12. Prazma J, Vance SG, Bolster ED, Pillsbury HC, Postma SD. Cochlear blood flow, the effect of noise at 60 minutes’ exposure. Arch Otolaryngol Head Neck Surg 1987; 113:36-39.
13. Quirk WS, Seidman MD. Cochlear vascular changes in response to loud noise. Am J Otol 1995; 16:322-325.
14. Krishnan A, Mattox DE, Fountain AJ, Hudgins PA. CT arteriography and venography in pulsatile tinnitus: preliminary results. AJNR Am J Neuroradiol 2006; 27:1635-1638.
15. Koyuncu M, Elhami AR, Akan H, Sahin M, Basoglu T, Simsek M. Investigation of the vertebrobasilar arterial system in vertigo by vestibulocochlear test, SPECT and angiography. Auris Nasus Larynx 2001; 28:23-28.
16. Nakashima T, Naganawa S, Sone M, Tominaga M, Hayashi H, Yamamoto H, et al. Disorders of cochlear blood flow. Brain Res Brain Res Rev 2003; 43:17-28.
17. Iwasaki S, Nagura M, Miyashita H, Umemura K, Hoshino T. Focal damage to cochlear microcirculation measured using a non-contact laser blood flowmeter in guinea pigs. Acta Otolaryngol 1998; 118:666-672. 18. Olszewski J, Chudzik W, Miłoński J, Kuśmierczyk K. Qualitative and quantitative studies in electron microscopy on influence of experimental ischemia of the vertebral arteries on the outer hair cells function in guinea pigs. Acta Otorhinolaryngol Belg 2003; 57:151-154.
19. Mom T, Telischi FF, Martin GK, Stagner BB, Lonsbury-Martin BL.Vasospasm of the internal auditory artery: significance in cerebellopontine angle surgery. Am J Otol 2000; 21:735-742.
20. Scheibe F, Haupt H, Baumgartl H. Effects of experimental cochlear thrombosis on oxygenation and auditory function of the inner ear. Eur Arch Otorhinolaryngol 1997; 254:91-94.
21. Morrison AW. Acute deafness. Br J Hosp Med 1978; 19:237-249.
22. Campbell G, Renner G, Estrem SA. Bilateral aberrant internal carotid arteries. Otolaryngol Head and Neck Surg 1992; 107:124-128.
23. Ciorba A, Bianchini C, Ortore RP, Bovo R, Martini A. Aberrant internal carotid artery in the middle ear: two case reports. B-ENT 2007; 3: 191-194.
24. Windfuhr JP. Aberrant internal carotid artery in the middle ear. Ann Otol Rhinol Laryngol Suppl. 2004; 192:1-16.
25. Parnes LS, Shmotakahara SG, Pelz D, Lee D, Fox AJ. Vascular relationships of the vestibulocochlear nerve on magnetic resonance imaging. Am J Otol 1990; 11:278-281.
26. Odkvist LM, Thuomas KA, Niklasson M. Macrovascular causes underlying otoneurological disturbances. Acta otolaryngol (Stockh) 1994; 115:145-148.
27. Chadha NK, Weiner GM. Vascular loops causing otological symptoms: a systematic review and meta-analysis. Clin Otolaryngol 2008; 33: 5-11.
28. Liu CH, Lin SK, Chang YJ. Cochlear vertebral entrapment syndrome: a case report. Eur J Radiol 2001; 40:147-150.
29. Rinehart R, Harre RG, Roski AR, Dolan KD. Aneurysm of the anterior inferior cerebellar artery producing hearing loss. Ann Otol Laryngol 1992; 101:705-706.
30. Sarkar A, Link MJ. Distal anterior inferior cerebellar artery aneurysm masquerading as a cerebellopontine angle tumor: case report and review of literature. Skull Base 2004; 14:101- 106.
31. Good CD, Phelps PD, Lim DP. Case report: greatly enlarged jugular fossa with progressive sensorineural hearing loss. J Laryngol Otol 1995; 109:350-352.
32. Haupert MS, Madgy DN, Belenky WM, Becker JW. Unilateral conductive hearing loss secondary to high jugular bulb in a pediatric patient. Ear-Nose-Throat J 1997; 76:468-469.
33. Bricoli A, De Micheli E, Gambin R, Alessandrini F, Iuzzolini P. Cavernous malformation of the internal auditory canal. A case report. J Neurosurg Sci 1995; 39:153-158.
34. Strome SE, Hartshorn DO, Carroll WR, Sheard N, Disher MJ. Otologic manifestations of vertebral artery dissection. Otolaryngol Head Neck Surg 1997; 116:234-237.
35. Nagahata M, Hosoya T, Fuse T, Aoyagi M, Yamaguchi K. Arterial dissection of the Nadir Yıldırım Van Tıp Dergisi, Cilt:19, Sayı:3, Temmuz/2012 156 vertebrobasilar systems: A possible cause of acute sensorineural hearing loss. Am J Otol 1997; 18: 26-31.
36. Horii A, Okumura K, Kitahara T, Kubo T. Intracranial vertebral artery dissection mimicking acute peripheral vertigo. Acta Otolaryngol 2006; 126:170-173.
37. Synder SO. Unilateral sudden hearing loss as a result of anomalous carotid anatomy. J Vasc Surg 1990; 12:341-344.
38. Pirodda A, Sorrenti G, Marliani AF, Cappello I. Arterial anomalies of the middle ear associated with stapes ankylosis. J Laryngol Otol 1994; 108: 237-239.
39. Umezu H, Seki Y, Aiba T, Kumakawa K. Aneurysm arising from the petrous portion of the internal carotid artery: case report. Radiat Med Med Imaging Radiat Oncol 1993; 11:251-255.
40. Kim JS, Lopez I, DiPatre PL, Liu F, Ishiyama A, Baloh RW. Internal auditory artery infarction: clinicopathologic correlation. Neurology 1999; 52:40-44.
41. Gross M, Banin E, Eliashar R, Ben-Hur T. Susac syndrome. Otol Neurotol 2004; 25: 470-473.
42. Eliashar R, Saah D, Osin P, Sichel JY. Hemangioendothelioma of the temporal bone in a child. Int J Pediatr Otolaryngol 1997; 76:468- 469.
43. Pappas DG, Schneiderman TS, Brackmann DE, Simpson LC, Chandra-Sekar B, Sofferman RA. Cavernous hemangiomas of the internal auditory canal. Otolaryngol Head Neck Surg 1989; 101: 27-32.
44. Roland PS, Glasscock III ME. Surgery of the cranial base in “Otolaryngology”. Eds: Paparella MM, Shumrick DA, Gluckman JL, Meyerhoff WL. Third ed. Saunders; Philadephia, PA 1991; 1789-1807
45. Megerian CA, McKenna MJ, Nadol JB.Jr. Nonparaganglioma jugular foramen lesions masquerading as glomus jugulare tumors. Am J Otol 1995; 16:94-98.
46. Hindersin S, Schubert O, Cohnen M, Felsberg J, Schipper J, Hoffmann TK. Angiosarcoma of the temporal bone. Laryngorhinootologie 2008; 87: 345-348.
47. Yamakami I, Ono JA. Sigmoid sinus dural arteriovenous malformation resulting from jugular foramen schwannoma–case report. Neurol Med Chir (Tokyo) 1998; 38:43-46.
48. Kido T, Sekitani T, Okinaki Y, Tahara T, Hara H. A case of cerebellar infarction occurred with the 8th cranial nerve symptoms. Auris-NasusLarynx (Tokyo) 1994; 21:111-117.
49. Lee H, Kim HJ, Koo JW, Kim JS. Progression of acute cochleovestibulopathy into anterior inferior cerebellar artery infarction. J Neurol Sci 2009; 278: 119-122.
50. Park JH, Kim H, Han HJ. Recurrent audiovestibular disturbance initially mimicking Ménière’s disease in a patient with anterior inferior cerebellar infarction. Neurol Sci 2008; 29:359-362.
51. Hinojosa R, Kohut RI: Clinical diagnosis of anterior inferior cerebellar artery trombosis. Ann Otol Rhinol Laryngol 1990; 99:261-271.
52. Baloh R.W. Vestibular disorders due to cerebrovascular disease in “Disorders of the Vestibular System”. Eds: Baloh RW, Halmagyi GM. Oxford University Press; New York, N.Y. 1996; 418-429.
53. Lee H, Baloh RW. Sudden deafness in vertebrobasilar ischemia: clinical features, vascular topographical patterns and long-term outcome. J Neurol Sci 2005; 228:99-104.
54. Sauvaget E, Kici S, Petelle B, Kania R, Chabriat H, Herman P, Tran Ba Huy P. Vertebrobasilar occlusive disorders presenting as sudden sensorineural hearing loss. Laryngoscope 2004; 114:327-332.
55. Lee H. Sudden deafness related to posterior circulation infarction in the territory of the nonanterior inferior cerebellar artery: frequency, origin. Eur Neurol 2008; 59:302-306.
56. Susmano A, Rosenbush SW. Hearing loss and ischemic heart disease. Am J Otol 1988; 9:403- 408.
57. McKennan KX, Nielsen SL, Watson C, Wiesner K. Meniere’s Syndrome: An atypical presentation of giant cell arteritis (Temporal arteritis). Laryngoscope 1993; 103:1103-1107.
58. Gates GA, Cobb JL, D`agostino RB, Wolf PA. The relation of hearing in elderly to the presence of cardiovascular disease and cardiovascular risk factors. Arch Otolarngol Head Neck Surg 1993; 119:156-161. 59. Liu F, Xia M, Xu A. Expression of VEGF, iNOS, and eNOS is increased in cochlea of diabetic rat. Acta Oto-Laryngologica 2008; 128: 1178-1186.
60. Esparza CM, Jáuregui-Renaud K, Morelos CM, Muhl GE, Mendez MN, Carillo NS, et al. Systemic high blood pressure and inner ear dysfunction: a preliminary study. Clin Otolaryngol 2007; 32:173-178.
61. Pirodda A, Brandolini C, Ferri GG, Modugno GC, Esposti DD, Borghi C. Inner ear dysfunction of uncertain origin: a multidisciplinary approach could give something more. Med Hypotheses 2009; 72: 188-189.
62. Axelsson A, Borg E, Hornstrand C. Noise effect on the cochlear vasculature in normotensive and spontaneously hypertensive rats. Acta Otolaryngol 1983; 96:215-225.
63. Yoon TH, Paparella MM, Schachern PA. Systemic vasculitis: a temporal bone histopathologic study. Laryngoscope 1989; 99: 600-609.
64. Per-Lee JH, Parsons R. Vasculitis presenting as otitis media. South Med J 1969; 62:161-165.
65. Rowe-Jones JM, Macallan DC, Sorooshian M. Polyarteritis nodosa presenting as bilateral sudden onset cochleo-vestibular failure in a young woman. J Laryngol Otol 1990; 104:562- 564.
66. Yildirim N, Arslanoglu A, Aygun N. Otologic Hearing in Vascular Disorders Derleme Van Tıp Dergisi, Cilt:19, Sayı:3, Temmuz/2012 157 and leptomeningeal involvements as presenting features in seronegative Wegener granulomatosis. Am J Otolaryngol 2008; 29:147- 149.
67. Cheson BD, Bluming AG, Alroy J. Cogan’s syndrome: Systemic vasculitis. Am J Med 1977; 65:549-555. 68. Selivanova O, Haxel BR, Mann WJ. Cogan’s syndrome: a diagnostic challenge. HNO 2006; 54: 619-623. 69. Amor-Dorado JC, Arias-Nuñez MC, MirandaFilloy JA, Gonzalez-Juanatey C, Llorca J, Gonzalez-Gay MA. Audiovestibular manifestations in patients with limited systemic sclerosis and centromere protein-B (CENP-B) antibodies. Medicine (Baltimore) 2008; 87:131- 141.
70. Hansen S. Postural hypotension–cochleovestibular hypoxia–deafness. Acta Otolaryngol (Stockh) 1988; 449:165-169.
71. Chao TK. Sudden sensorineural hearing loss after rapid reduction of blood pressure in malignant hypertension. Ann Otol Rhinol Laryngol 2004; 113:73-75.
72. Misrahy GH, Shinabarger EW, Arnold JE. Changes in cochlear endolymphatic oxygen availability, action potential, and microphonics during and following asphyxia, hypoxia and exposure to loud sounds. J Acoust Soc Am 1958; 30:701-704.
73. Nuttall AL, Hultcrantz E, Lawrence M. Does loud sound influence the intracochlear oxygen tension. Hear Res 1981; 5:286-293.
74. Tyagi I, Tsutomu N, Ito A, Yanagita N. Effect of hemorrhagic hypotension on blood flow to the basal and upper turns of the cochlea. AurisNasus-Larynx (Tokyo) 1995; 22:93-95.
75. Yamamoto H, Makimoto K. Sensitivity of the endocochlear potential level to cochlear blood flow during hypoventilation. Ann Otol Rhinol Laryngol 2000; 109:945-951.
76. Hall SJ, McGuigan JA, Rocks MJ. Red blood cell deformability in sudden sensorineural deafness: another aetiology? Clin Otolaryngol 1991; 16:3-7.
77. Browning GG, Gatehous S, Lowe GDO. Blood viscosity as a factor in sensorineural hearing impairment: Lancet 1986; 18:121-123.
78. Burch-Sims GP, Matlock VR. Hearing loss and auditory function in sickle cell disease. J Commun Disord 2005; 38:321-329.
79. Nakai Y, Masutani H. Noise-induced vasoconstriction in the cochlea. Acta Otolaryngol (Stockh) 1988; Suppl.447:23-27.
80. Attanasio G, Buongiorno G, Piccoli F, Mafera B, Cordier A, Barbara M, et al. Laser Doppler measurement of cochlear blood flow changes during conditioning noise exposure. Acta Otolaryngol 2001; 121:465-469. 81. Wright JW, Dengerink HA, Miller JM, Goodwin PC. Potential role of angiotensin II in noiseinduced increases in inner ear blood flow. Hearing Research 1985; 17:41-46.
82. Miller JM, Brown JN, Schacht J. 8-isoprostaglandin F (2alpha), a product of noise exposure, reduces inner ear blood flow. Audiol Neurootol 2003; 8:207-221.
Interruzione del flusso di sangue “Dr. Travis Moore westib.com.”
Questa sezione esamina le conseguenze che sorgono quando che il flusso di sangue è compromesso. Prima si vedranno i danni in gran parte reversibili che possono verificarsi a seguito di ischemia transitoria – una riduzione del flusso di sangue adeguato al tessuto. Successivo faremo affrontare i danni a causa di cessazione del flusso di sangue che dura a causa del blocco di una nave; questo grave, ischemia prolungata porta a “infarto”. Per ultimo verrà affrontata un altro tipo di grave ischemia: l’emorragia.
Ischemia e l’attacco ischemico transitorio (TIA)
In media, qualcuno ha un ictus negli Stati Uniti ogni 40 secondi. 1 La prevalenza della malattia vascolare è troppo grande per essere familiarità con i suoi principi fondamentali. In effetti, uno studio svedese ha scoperto che circa 1/4 dei casi di vertigine acuta isolata potrebbe essere attribuito ad caudale infarto cerebellare. 2 E ‘importante notare partecipanti allo studio sono stati ricoverati in una clinica di emergenza, il che significa audiologi e gli altri medici non esercitano in tale un ambiente è improbabile incontrare questi pazienti inizialmente. Tuttavia, questa informazione è utile per evitare la diagnosi non corretta di un disturbo labirintica durante l’esame vestibolare. Mentre il labirinto può infatti essere influenzata, il punto dello studio sopra menzionato è che la vera eziologia può benissimo essere di natura vascolare.
Andiamo a scavare definendo due termini importanti:
Ischemia
Fornitura carente di sangue a una parte del corpo (come il cuore o il cervello) che è causa di ostruzione del flusso di sangue arterioso (come dal restringimento delle arterie di spasmo o malattie). 3
Attacco ischemico transitorio
Un breve episodio di ischemia cerebrale che di solito è caratterizzata da oscurare temporaneo di vista, slurring di parola, intorpidimento, paralisi, o sincope e che è spesso predittivo di un grave ictus, TIA sigla; chiamato anche mini-ictus. 3
I segni e sintomi neurologici di un TIA sono generalmente di breve durata, senza effetti duraturi. TIA sono comuni negli anziani, e sono spesso indicativi di più gravi eventi ischemici a venire. 1 4 Per illustrare l’importazione di una breve timecourse per il recupero da un evento ischemico, consideriamo il labirinto. Ricordiamo che l’orecchio interno è particolarmente suscettibile di interruzioni nel flusso sanguigno come viene fornito da una singola arteria senza fine collaterali (labirintico / uditivo interno). Un altro fattore che rende il labirinto altamente suscettibile di eventi ischemici è il metabolismo ad alta energia del orecchio interno. 4 Questa sensibilità è stata approssimativamente quantificato in studi su animali, con i risultati tipici che mostrano potenziali cocleari decrescenti entro un minuto di ischemia. Funzione cocleare generale, viene recuperata se l’ischemia dura meno di 10 minuti. Periodi senza il flusso di sangue che dura 15 a 30 minuti si traducono in perdita di cellule ciliate esterne e danno cocleare permanenti. 5 Tenete a mente diversi tessuti hanno diverse soglie di tempo per indurre disfunzioni e necrosi del tessuto in modo che i tempi di cui sopra si applicano specificamente per l’orecchio interno, ma la modello generale è universale (cioè, più breve è l’evento ischemico migliori sono le possibilità di recupero).
Con un po ‘di storia sotto le nostre cinture, ora consideriamo un altro disturbo vascolare importante:
Insufficienza vertebro-basilare (VBI) 6
Insufficienza vertebro-basilare (VBI) è una causa molto comune di vertigini a causa di ischemia, soprattutto nella popolazione anziana. I sintomi più comuni sono descritti di seguito:
Sintomi
· esordio improvviso
· alcuni minuti di durata
· vertigine
· nausea e vomito
· difetti del campo visivo *
· diplopia *
· mal di testa *
· * I sintomi causati da ischemia del circolo posteriore
Notare che i sintomi sono insorgenza improvvisa; questo è altamente tipico di tutte le lesioni vascolari. Non ci vuole molto per tessuto neurale sensibile a reagire da inadeguato flusso sanguigno. Confrontare questo alla comparsa più insidioso di sintomi causati da tumori a crescita lenta. Così referto del paziente di insorgenza improvvisa di solito esclude neoplasie e punti invece di una eziologia vascolare.
VBI può produrre vertigini da solo come un sintomo precoce, ma alla fine sarà accompagnato da altri sintomi circolo posteriore. Infatti, episodi di vertigine consistenti senza l’eventuale comparsa degli altri sintomi sopra elencati sono indicativi di qualcosa di diverso VBI.
I sintomi possono essere causate da aterosclerosi della succlavia, vertebrale, o arterie basilari. L’aterosclerosi si riferisce alla formazione di placca grassi all’interno arterie, che alla fine si indurisce e restringe i vasi sanguigni. I sintomi sono anche precipitate da furto della succlavia, anche se questa condizione è rara. Subclavian Steal risultati sindrome di VBI quando il sangue viene travasato lungo l’arteria vertebrale, lontano dal circolo posteriore, per fornire alle estremità superiori. Comprensibilmente in questo caso, VBI e suoi sintomi associati sarebbero stati portati sulla esercitando le braccia e le spalle. Ischemia del circolo posteriore conseguente VBI può essere causato anche semplicemente “guardando” in alcuni pazienti. Cioè, quando inarcando il collo posteriore è possibile “nodo” dell’arteria basilare. Ciò può verificarsi da spondilosi cervicale, una malattia degenerativa della colonna vertebrale. Altri fattori scatenanti possono essere dovute a ipotensione ortostatica o Stokes Adams attacchi che sono discussi nella sezione sulla pressione sanguigna.
Il più delle volte, il trattamento di insufficienza vertebro-basilare si fonda sul controllo dei fattori di rischio associati con la malattia vascolare. Condizioni mirate comuni includono:
· diabete mellito
· ipertensione
· iperlipidemia
Farmaci antiaggreganti piastrinici, come l’aspirina, sono spesso efficaci nei casi più lievi. Nei casi più gravi farmaci anticoagulanti, come il warfarin / Coumadin, può essere necessaria. Questo è soprattutto il caso se la trombosi dell’arteria basilare è sospettata. Impareremo di più su trombi più in basso nella pagina.
Infarto e ictus ischemico
La parola “infarto” è riconoscibile per molti dalla vita quotidiana. Il termine “infarto del miocardio”, riferendosi a un attacco di cuore è diventato volgare abbastanza popolare. Tuttavia, che cosa è esattamente un infarto? Le definizioni di seguito riportati chiarire la situazione prima di passare.
Infarto
Un’area di necrosi in un tessuto o organo risultante dalla ostruzione della circolazione locale da un trombo o embolia. 3
Infarto
Il processo di formazione di un infarto. 3
Un infarto è danneggiato tessuto da una mancanza di flusso sanguigno. È importante comprendere il rapporto tra infarto e ischemia (di cui sopra). Vale a dire, infarto si verifica a causa di ischemia, o in altre parole, il tessuto muore quando non riceve la corretta irrigazione. Infarto si può verificare da ischemia causata da due tipi di ostruzione vascolare: un trombo o di un embolo. Un trombo è più comunemente noto come un coagulo di sangue, ed è collegata a parete di un vaso sanguigno. Viceversa, “embolo” si riferisce a una particella che circola attraverso il sangue. Questi tipi di occlusione arteriosa sono l’eziologia dietro “ictus ischemico”, che rappresenta il 87% di tutti gli ictus. 4
Embolo
Una particella anormale (come una bolla d’aria) che circola nel sangue. 3
Trombo
Un coagulo di sangue formato all’interno di un vaso sanguigno e rimanendo attaccato al suo luogo di origine.3
Neurologi treno per anni per determinare il luogo di blocco dei vasi considerando la mappa intricata delle arterie e delle strutture quelli arterie fornitura. Mentre questa comprensione in profondità e abilità diagnostica è ben fuori della portata di questo sito web, noi affronteremo alcune grandi considerazioni immagine site-of-lesione che permetterà di fare luce su comuni (e alcuni non così comuni) disturbi dell’equilibrio. Abbiamo già discusso il pertinente sistema vascolare delle strutture di equilibrio, e Esaminiamo ora i vari siti di ostruzione arteriosa e come il conseguente infarto può influenzare i vostri pazienti.
Infarto del tronco cerebrale 6
Le Sindromi associate con l’ictus della parte dell’arteria cerebellare posteriore inferiore (PICA) e dell’arteria cerebellare antero inferiore (AICA) sono comuni, e provocano danni al tronco cerebrale e / o al cervelletto. Le tabelle che seguono riassumono i principali segni e sintomi prodotti da occlusione della PICA e AICA. 6
TABELLA 1.
|
Sintomi e segni |
PICA Infarct |
|
Vertigo, nistagmo |
Nuclei vestibolari, posteriore inferiore del cervelletto |
|
Tinnito, perdita dell’udito |
N / A |
|
Andatura e degli arti atassia |
Ventrale tratto spinocerebellare, posteriore inferiore del cervelletto |
|
La disfagia, diminuzione bavaglio |
Nuclei vagali e nervi |
|
Hemianesthesia viso |
CN V e nucleo |
|
Paralisi facciale |
CN VII |
|
Perdita hemisensory Crossed |
Tratto spinotalamico |
|
Sindrome di Horner |
Scendendo fibre simpatiche |
Si può notare che le vertigini causate da un blocco della PICA seguono l’alimentazione dell’arteria: per esempio, i nuclei vestibolari risiedono nel midollo del tronco cerebrale, che viene fornito dalla PICA. Di conseguenza, le vertigini sono il prodotto dalla lesione dei nuclei vestibolari e / o parte del cervelletto inferiore – non interessando direttamente l’apparato vestibolare / orecchio interno. Anche nel midollo si trova è il nucleo del X NC, il nervo vago. Danni a questo nervo possono portare a disfagia e ad un riduzione del riflesso del vomito Sintomi uditivi sono assenti, come ancora una volta, il labirinto è inalterato.
Atassia
L’incapacità di coordinare i movimenti volontari muscolari che è sintomatico di alcuni disturbi nervosi. 3
Disfagia
Difficoltà a deglutire. 3
Sindrome di Horner
![]()
“Miosis” di Waster – Nautiyal A, Singh S, DiSalle M, O’Sullivan J (2005)
Dal lato in cui il sistema nervoso è stato leso si noteranno:
· Restringimento della rima palpebrale superiore per paralisi del muscolo tarsale superiore (simile ma non uguale alla ptosi palpebrale, ossia un abbassamento della palpebra più marcato, legata alla paralisi del nervo oculomotore che innerva il muscolo elevatore della palpebra superiore)
· Enoftalmo, ossia il rientro dell’occhio nell’orbita (spesso non presente)
· Miosi, ossia restringimento della pupilla
· Anidrosi, ossia l’assenza di sudorazione, sul volto in questo caso (presente solo se la lesione avviene inferiormente alla biforcazione carotidea. Superiormente le fibre simpatiche deputate alla sudorazione seguono l’arteria carotide esterna, le rimanenti l’arteria carotide interna) 3
TABELLA 2.
|
Sintomi e segni |
AICA Infarct |
|
Vertigo, nistagmo |
Labirinto, nervo vestibolare, flocculo |
|
Tinnito, perdita dell’udito |
Coclea, nervo acustico, nucleo cocleare |
|
Andatura e degli arti atassia |
Medio peduncolo cerebellare, anteriore inferiore del cervelletto |
|
La disfagia, diminuzione bavaglio |
Nessuna |
|
Hemianesthesia viso |
CN V e nucleo |
|
Paralisi facciale |
CN VII |
|
Perdita hemisensory Crossed |
Tratto spinotalamico |
|
Sindrome di Horner |
Scendendo fibre simpatiche |
In contrasto tabella 2 con infarto a causa dell’occlusione lungo il PICA. Mentre le vertigini sono ancora il sintomo importante da occlusione dell’AICA, la sua eziologia è cambiata. Si ricorda che l’infarto dell’ AICA spesso danneggia l’arteria labirintico, che è il solo apporto di sangue ossigenato all’orecchio interno. Così, vertigini precipita causa lesione del labirinto (e / o il nervo vestibolare e flocculo cerebellare). Ciò significa che l’ ipoacusia e / o gli acufeni (cioè, sintomi uditivi) sono presenti insieme alle vertigini perché l’apparato vestibolare, coclea e CN VIII sono tutti fornite dal AICA attraverso l’arteria labirintica. Inoltre, la disfagia e riflesso faringeo diminuito o o assente, come la AICA non fornisce il midollo.
Queste differenze nei sintomi sulla base di quali strutture ogni arteria forniture ci permette di fare alcune conclusioni generali sul sito di lesione. Prendiamo in considerazione queste differenze di due sindromi comuni causate da occlusione del PICA e AICA.
Sindrome di Wallenberg (sindrome di Wallenberg) 6 7
Sindrome di Wallenberg è una condizione molto comune, con conseguente infarto del midollo laterale. Anche se questo può portare all’assunzione del vaso occluso è il PICA, questo è raramente il caso; invece, l’arteria vertebrale è tipicamente il sito di lesione. Tuttavia, questo provoca ancora miocardio delle strutture forniti dal PICA, perché il PICA nascono dalle arterie vertebrali (vedi figura 1). Ci sono parecchi sintomi e segni di infarto midollare laterale caratteristiche:
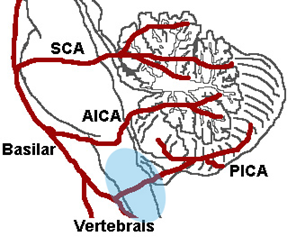
FIGURA 1 Wikimedia Commons
Sintomi
· vertigine
· diplopia
· disfasia
· disfonia
· dolore facciale omolaterale
Segni
· sindrome di Horner omolaterale
· perdita del dolore e della temperatura sul lato lesionato della faccia
o (V CN nervi e nucleo)
· la paralisi omolaterale della gola
o (CN IX e CN X)
· retto laterale omolaterale e debolezza facciale
· dismetria omolaterale, aritmia e dysdiadochokinesia
o (Cervelletto)
· nistagmo spontaneo
o (Nucleo vestibolare)
· perdita di dolore e temperature sensazione sul lato del corpo opposta della lesione
o (Attraversato tratti spinotalamico)
Questo suona familiare? Questi segni e sintomi sono gli stessi di quelli elencati nella tabella 1 di cui sopra (PICA Infarto).Questo dovrebbe guidare a casa che la conoscenza dei principali vasi e quello dei tessuti che forniscono possono essere estremamente utile nella diagnosi differenziale. Tenere presente non tutti questi sintomi saranno presenti in ogni paziente, come la gravità della sindrome si basa dall’entità dei tessuti danneggiati (cioè, la dimensione e la posizione della lesione). Alcuni pazienti avvertiranno diminuendo i sintomi in poche settimane o mesi (meno comuni), mentre altri sperimenteranno difficoltà per anni (più comune). Sia diagnosi e la prognosi sono lasciati il neurologo, ma riconoscere questi segni e sintomi possono rivelarsi utili nel predire e interpretare i propri risultati del test.
Sindrome Laterale Pontomidollare 6
Come abbiamo appreso in precedenza, l’occlusione della AICA può influenzare la zona pontomidollare e parti del cervelletto inferiore. Il peduncolo cerebellare medio è spesso la zona più colpita. Come visto sopra, con la sindrome di Wallenberg sopra, i segni ed i sintomi della sindrome pontomidollare laterale sono facili da intuire in base alle strutture fornite dal AICA (vedi figura 2).
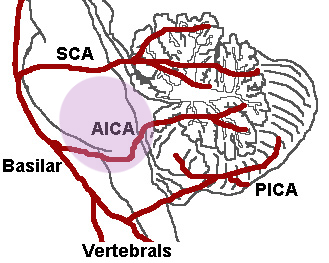
FIGURA 2 Wikimedia Commons
Sintomi
· vertigini grave
· nausea e vomito
· perdita uditiva unilaterale
· tinnito
· paralisi facciale
· scarsa coordinazione (asinergia) del cervelletto
Segni
· perdita dell’udito omolaterale e / o tinnito
o (labirinto e porzione uditiva del CN VIII)
· nistagmo spontaneo
o (labirinto e parte vestibolare CN VIII)
· la debolezza ipsilaterale del viso
o (CN VII e nucleo)
· cerebellare disfunzione omolaterale
o (anteriore inferiore del cervelletto)
· perdita omolaterale del dolore e della temperatura sensazione sulla faccia
o (CN V e nucleo)
· perdita controlaterale del dolore e della temperatura senstion sul corpo
o (attraversato fibre spinotalamico)
Come descritto in precedenza, si è visto che si possono instaurarsi sofferenze cocleari con occlusione dell’ AICA, e la causa della vertigine è labirintica. Mentre gli effetti residui possono gradualmente migliorare nel tempo, la vertigine può durare per settimane o addirittura mesi a causa di danni di compensazione centrali strutture.
Il cervelletto gioca un ruolo fondamentale nel controllo motorio e la tempistica, avendo effetti profondi sui movimenti oculari, la parola, i movimenti degli arti, la postura e l’andatura. Si può imparare tutto sul cervelletto visitando la sua sezione di questo sito web. Per ora, l’attenzione sarà concentrata sulle conseguenze di infarto cerebellare.
Per iniziare, diamo esporre comportamento potenzialmente ingannevole di ischemia cerebellare: infarto può essere isolato al cervelletto, senza influenzare il tronco cerebrale. Come è ingannevole? Prendere in considerazione i sintomi di infarto isolato al cervelletto:
Sintomi
· vertigini grave
· vomito
· atassia
Questi sintomi, senza la presenza di segni del tronco cerebrale (di cui sopra), può essere confuso con un episodio periferico / labirintico acuta. Caratteristiche che aiutano a identificare infarto cerebellare come tale includono andatura atassia, arti atassia, e lo sguardo-evocato (gaze paretica) nistagmo. Naturalmente il metodo migliore per rivelare infarto cerebellare è con una risonanza magnetica.
Con sopra avvertimento in mente, torniamo indietro a potenziali fonti di ischemia. Se vi ricordate le arterie del circolo posteriore, le seguenti navi non sono una sorpresa:
Arterie di infarto cerebellare isolato
· arteria vertebrale
· posterior inferiore arteria cerebellare (PICA)
· anteriore inferiore arteria cerebellare (AICA)
· arteria cerebellare superiore (SCA)
Come discusso in precedenza, l’occlusione degli stessi vasi può anche portare a tronco cerebrale infarto. Quando una lesione colpisce sia il tronco cerebrale e cervelletto, entrambi i tipi di segni e sintomi saranno presenti. Clinicamente, è facile immaginare la potenziale gravità di un tale evento.
Un altro punto da tenere a mente è il potenziale di gonfiore cerebellare in seguito ad un attacco ischemico. Dopo un periodo di latenza di 24 a 96 ore, è possibile per il cervelletto diventi abbastanza gonfio per iniziare causando segni tronco cerebrale progressiva. Il risultato, se non controllata, porta alla tetraplegia, coma, poi la morte. Chirurgia di decompressione chirurgica è necessaria per alleviare questa condizione.
Infarto del Labirinto 6
La familiarità con la fornitura di sangue all’orecchio interno è essenziale per comprendere le sindromi ictus che colpiscono il labirinto. Tuttavia, proprio come con il resto del circolo posteriore, dopo l’apporto di sangue è padroneggiata i segni ed i sintomi seguono naturalmente e sono facili da ricordare. Tenete a mente quanto segue di solito si verifica nella popolazione anziana con una storia di malattia vascolare, ictus, TIA, ecc
Perdita uditiva unilaterale improvvisa e vertigini gravi possono essere causati da ischemia dell’arteria labirintica. Poiché l’occlusione si sarebbe verificato prima che le divisioni delle arterie, entrambe le strutture vestibolari e cocleari sono interessati.
Improvvisa perdita dell’udito senza sintomi vertiginosi può essere attribuito alla ischemia dell’arteria cocleare comune o uno dei suoi rami.
Vertigine senza perdita dell’udito può essere dovuto a infarto del labirinto vestibolare a causa l’occlusione della arteria vestibolare anteriore. La vertigine può durare minuti o addirittura giorni a seconda della gravità della lesione. La descrizione clinica è “improvviso, grave vertigini senza sentire la perdita o del tronco cerebrale sintomi”. 6
Anche se benigna vertigine parossistica posizionale (VPPB) è analizzato approfonditamente altrove , vale la pena accennare qui, come BPPV è una possibile sequela a seguito di un attacco ischemico che coinvolge l’arteria vestibolare anteriore. Infarto della macula del otricolo può causare il rilascio del suo otoconia nel canale semicircolare posteriore. Il canale posteriore può ancora generare vertigini a causa del fatto che può essere ancora intatto, come fornito dal arteria cocleare comune.
Bibliografia
Baloh, R., & Honrubia, V. (2001). Clinical neurophysiology of the vestibular system. (3rd ed., Vol. 63). New York: Oxford University Press.
Kim, J. S., Lee, H. (2009). Inner Ear Dysfunction Due to Vertebrobasilar Ischemic Stroke. Seminars in Neurology, 29(5), 534-540.
Lasslo, R. (1997). Theory of eye motion and neurological testing. Retrieved from http://cim.ucdavis.edu/eyes/version1/eyeText.htm
Manto, M., et al. (2011). Consensus Paper: Roles of the Cerebellum in Motor Control – The Diversity of Ideas on Cerebellar Involvement in Movement. The Cerebellum.
Merriam-Webster Medical Dictionary [Internet]. [Springfield (MA)]: Merriam-Webster, Incorporated; ©2003. Available from:
http://www.nlm.nih.gov/medlineplus/mplusdictionary.html.
Montgomery, T. (2013). The extraocular muscles. Retrieved from http://www.tedmontgomery.com/the_eye/eom.html
NINDS Neuroleptic malignant syndrome information page. (2007, February 14). Retrieved from
http://www.ninds.nih.gov/disorders/neuroleptic_syndrome/neuroleptic_syndrome.htm
NINDS Wallenberg’s syndrome information page. (2007, February 15). Retrieved from http://tinyurl.com/mqjrund
Norrving, B., Magnusson, M., & Holtas, S. (1995). Isolated acute vertigo in the elderly; vestibular or vascular disease? Acta Neurol Scand, 91(1), 43-48.
O’Rourke, R. A. (1972). Clinical cardiology: The stokes-adams syndrome – definition and etiology; mechanisms and treatment. California Medicine, 117(1), 96-99. Retrieved from http://tinyurl.com/lh2hh8e
Pychiatric Nursing. (2012, April 14). Neuroleptic-induced extrapyramidal symptoms. Retrieved from
http://nursingplanet.com/psychopharmacology/extrapyramidal_symptoms.html
Rosamond, W., Flegal, K., Furie, K., et al. (2008). Heart Disease and Stroke Statistics−−2008 Update : A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation, 117, e25-e146.
Takeda, N., Morita, M., & Hasegawa, S., et al. (1993). Neuropharmacology of motion sickness and emesis. Acta Otolaryngol (Stockh), 510, 10-15.
Understanding blood pressure readings. (2012, April 04). Retrieved from http://tinyurl.com/34kf568 (American Heart Association).
Young, P. A., Young, P. H., & Tolbert, D. (2008). Basic clinical neuroscience. (2nd ed.). Wolters-Kluwer Health: Philadelphia.
ESAMI STRUMENTALI
Indagini audiometriche e vestibolari: la batteria di test audiometrici inizialmente proposta prevede un esame audiometrico tonale liminare (alle frequenze 125, 250, 500, 1000, 2000, 4000, 8000 Hz secondo l’International Organization for Standardization, ISO), audiometria vocale, audioimpedenzometria, potenziali evocati uditivi. Si può ampliare l’indagine ricorrendo alla ricerca delle emissioni otoacustiche e all’esecuzione dell’elettrococleografia. L’esame audiometrico tonale liminare deve porre in luce la presenza di un’ipoacusia di percezione che deve essere >30 dB e interessare almeno 3 frequenze consecutive. Questo costituisce la diagnosi fondamentale dell’ipoacusia improvvisa. Per valutare l’entità della perdita uditiva si usa solitamente la seguente classificazione: lieve, > 20 a ≤40 dB HL; moderata, > 40 a ≤ 70 dB HL; severa, > 70 a ≤ 90 dB HL; e profonda, > 90 dB HL. Soprattutto nelle forme profonde i residui uditivi fanno sì che il paziente spesso lamenti la percezione di un suono gracchiante e distorto. L’ipoacusia è monolaterale nel 95-98% dei casi. L’importanza maggiore della perdita uditiva e l’eventuale bilateralità sono considerati indici prognostici negativi. La bilateralità dell’ipoacusia deve far sospettare la presenza di lesioni centrali neoplastiche, infettive o correlate a sindromi paraneoplastiche o patologie autoimmuni. Non va sottovalutata in questi casi anche la possibilità di una patologia psichiatrica. Un’ipoacusia improvvisa di breve durata può anche far seguito ad un improvviso calo della pressione endocranica in seguito a punture lombari per prelievo di liquido cefalorachidiano o a interventi neurochirurgici. Nei casi di sordità con marcata caduta sulle frequenze gravi può essere considerata l’opportunità di un test con glicerolo o con diuretico al fine di svelare eventuali casi correlabili a idrope endolinfatica. Non si deve infatti dimenticare che almeno un 10% di paziente affetti da sindrome di Ménière presentano come prima manifestazione una ipoacusia improvvisa.
L’esame audiometrico tonale liminare costituisce anche la base del monitoraggio del soggetto nelle fasi evolutive del trattamento instaurato dopo la fase diagnostica. Solitamente i test audiometrici sono ripetuti quotidianamente per i primi 5 giorni e poi a giorni alterni per altri 5-10 giorni per monitorare l’evoluzione della malattia e l’efficacia della terapia. I controlli proseguono poi ad intervalli più ampi nei primi due mesi dall’attacco acuto.
La valutazione audiologica iniziale prosegue con lo studio del timpanogramma, la ricerca dei riflessi stapediali, l’audiometria vocale, un esame ABR ed un esame vestibolare. In casi particolari si può pensare di completare le indagini audiologiche con lo studio delle otoemissioni e/o elettrococleografia, ma questi due esami vengono effettuati solo in casi particolari. Tutto ciò può avere un valore orientativo nella diagnosi differenziale con patologie dell’orecchio medio e/o retrococleari. In particolare possono fornire le prime indicazioni di una patologia a carico del pacchetto acustico-facciale che sarà poi eventualmente confermata dall’imaging (RMN con e senza contrasto). Ricordiamo infatti che più del 10% dei casi di neurinoma dell’VIII nervo cranico hanno come prima manifestazione una ipoacusia improvvisa, ma solo 1-2 % delle ipoacusie improvvise è riconducibile ad un neurinoma (2,1% nella nostra casistica), anche perché la perdita uditiva può essere lenta e progressiva.
Nei casi in cui sia presente una sintomatogia suggestiva per una compartecipazione vestibolare si impone lo studio della funzionalità vestibolare che generalmente comprende la ricerca del nistagmo spontaneo e provocato, deviazioni segmentarie-toniche, ENG con termo stimolazioni e, ove possibile, studio dei VEMPs (vestibular-evoked miogenic potential).
Va però sottolineato come alcuni autori suggeriscano di eseguire tali test:
– a scopo diagnostico, anche in assenza di manifestazioni di sofferenza vestibolare perché possono svelare un deficit vestibolare asintomatico al momento dell’insorgenza della sintomatologia cocleare (ad esempio nell’ambito della prima manifestazione di un’idrope endolinfatica);
– a scopo prognostico perché l’evidenza di una sofferenza vestibolare associata all’ipoacusia improvvisa aggrava la prognosi. Allo stesso modo secondo alcuni AA viene attribuito un significato prognostico positivo del recupero uditivo del paziente al rilievo di normali risposte VEMPs.
In caso di sospetta fistola perilinfatica si può verificare semplicemente in videonistagmoscopia la comparsa di nistagmo in seguito a compressione pneumatica del timpano. Alcuni Autori hanno affinato tale test abbinandolo alla stabilometria.
Neuroimaging: basata sull’impiego della tomografia computerizzata (TC) e ancor meglio della risonanza magnetica nucleare (RM) contrastata con gadolinio è irrinunciabile nell’inquadramento diagnostico anche nei casi di completo e rapido recupero. Test MRI del cervello e IAC, test MRI del cervello rileva tumori così come fistola liquorale e stroke. Tumori, come ad esempio un neuroma acustico, può anche causare SHL che si risolve completamente (Nageris e Popovtzer, 2003). Test MRI non è sempre ordinata – in realtà, solo circa la metà del tempo (Coelho et al, 2011).
Esami di laboratorio. Le indagini di laboratorio vanno pianificate in stretta correlazione con la sintomatologia lamentata dal paziente, la storia clinica e di conseguenza con le cause eziologiche sospette. Possono includere:
– valutazione degli indici di flogosi (emocromo con formula leucocitaria, VES, PCR, o meglio PCR ultrasensibile).
– valutazione di eventuali emopatie o malattie metaboliche (studio completo della coagulazione, della funzionalità tiroidea, epatica e renale, trigliceridemia, colesterolemia, glicemia, omocisteinemia, vit B12, folati, AST, ALT yGT, elettroliti).
– valutazione di eventuali patologie autoimmuni o sindromi para-neoplastiche (ANA, ANCA, ENA, anticitocromo, antimitocondrio, antineurone, anticitrullina, anticorpi anticardiolipina, crioglobuline, anticoagulante lupico, trasformazioni linfocitarie).
– test sieroconversione per antigeni virali e/o batterici (Herpes simplex tipo 1, Cytomegalovirus, influenza e parainfluenza, EpsteinBarr virus, Coxsackievirus, hepatitis B e C virus, Toxoplasma gondii, screening malattie veneree, sifilide in particolare).
In casi particolari ed in presenza di un forte sospetto clinico, si possono aggiungere una serie di esami volti alla valutazione del rischio trombotico:
– PT, INR, PTT, fibrinogeno, D-Dimero;
– ricerca mutazione G1691A del Fattore V di Leiden;
– ricerca mutazione G677T del gene MTHFR;
– ricerca mutazione G20210A della protrombina;
– resistenza alla proteina C attivata (APCr);
– antitrombina 3
– autoanticorpi IgG e IgM antiprotrombina, anticardiolipina, anti beta 2 glicoproteina;
– proteine C ed S
– proconvertina.
Terapia. È Uno dei temi più controversi, soprattutto per quanto concerne la forma idiopatica. Tra i numerosi trattamenti proposti i glucocorticoidi sono, in tutto il mondo, generalmente i più adottati. In letteratura la maggioranza dei lavori riguarda la terapia cortisonica adottata da sola o in associazione con altri trattamenti per la cura delle ipoacusie improvvise, qualsiasi ne sia la natura supposta o accertata (diuretici, camera iperbarica, antiossidanti, anti aggreganti, vasoattivi). L’uso degli steroidi trova indicazione non solo nel trattamento della perdita uditiva, ma anche dell’acufene eventualmente ad essa associato. Studi condotti comparando gli effetti della carbamazepina e degli steroidi sugli acufeni non hanno dimostrato differenze significative.
La risoluzione spontanea è possibile in un grande numero di casi e avviene generalmente nei primi 15 giorni dall’instaurarsi del problema. Allo stesso modo un numero cospicuo di autori ha dimostrato come i migliori risultati con il trattamento corticosteroideo si verificano se questo è instaurato il prima possibile o quanto meno entro 1-2 settimane dalla comparsa dei sintomi. Minori risultati si hanno qualora il trattamento inizi 2 o più settimane dopo la comparsa della sintomatologia. La somministrazione sistemica, per os, intramuscolo o e.v., deve avvenire per non meno di 7 giorni e con un dosaggio di 1 mg/kg/die in caso di prednisone o equivalenti, e di 0,1 mg/kg/die di desametasone o equivalenti. Non sono note differenze significative tra le vie di somministrazione, molto più
importanti sono il dosaggio e la durata. In caso di terapia intratimpanica si consigliano almeno 5 somministrazioni desametasone alla concentrazione di 24 mg per ml. La terapia intratimpanica può precedere, accompagnare o seguire la somministrazione sistemica (6,18-33).
Bibliografia
27) Nakashima T, Naganawa S, Sone M, Tominaga M, Hayashi H, Yamamoto
H, Liu X, Nuttall AL. Disorders of cochlear blood flow. Brain Res Brain Res Rev. 2003 Sep;43(1):17-28.
28) Kariya S, Cureoglu S, Fukushima H, Morita N, Baylan MY, Maeda Y,
Nishizaki K, Paparella MM. Comparing the cochlear spiral modiolar artery in type-1 and type-2 diabetes mellitus: a human temporal bone study. Acta Med Okayama. 2010 Dec;64(6):375-83.
29) Weiss G, Werner-Felmayer G, Werner ER, Grünewald K, Wachter H,
Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med. 1994 September 1; 180(3): 969–976.
30) Wolff NA, Liu W, Fenton RA, Lee WK, Thévenod F, Smith CP. Ferroportin
1 is expressed basolaterally in rat kidney proximal tubule cells and iron excess increases its membrane trafficking. J Cell Mol Med. 2011 Feb;15(2):209-19.
31) Dai M, Yang Y, Omelchenko I, Nuttall AL, Kachelmeier A, Xiu R, Shi X.
Bone Marrow Cell Recruitment Mediated by Inducible Nitric Oxide Synthase/Stromal Cell-Derived Factor-1α Signaling Repairs the Acoustically Damaged Cochlear Blood-Labyrinth Barrier. Am J Pathol. 2010 Dec;177(6):3089-99.
46) ![]() kouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron
kouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron
transport and the role of transferring. Biochim Biophys Acta. 2012 Mar;1820(3):188-202.
47) Buretić-Tomljanović A, Vraneković J, Rubeša G, Jonovska S, Tomljanović
D, Sendula-Jengić V, Kapović M, Ristić S. HFE mutations and transferrin C1/C2 polymorphism among Croatian patients with schizophrenia and schizoaffective disorder. Mol Biol Rep. 2012 Mar;39(3):2253-8.
48) Babitt JL, Lin HY. The molecular pathogenesis of hereditary hemo‑
chromatosis. Semin Liver Dis. 2011 Aug;31(3):280-92.
49) Parajes S, González-Quintela A, Campos J, Quinteiro C, Domínguez F,
Loidi L. Genetic study of the hepcidin gene (HAMP) promoter and functional analysis of the c.-582A > G variant. BMC Genet. 2010 Dec 10;11:110.
50) Gemmati D, Zeri G, Orioli E, De Gaetano FE, Salvi F, Bartolomei I,
D’Alfonso S, Asselta R, Dall’Osso C, Leone MA, Singh AV, Zamboni P. Iron Gene Polymorphisms are Associated to Disability, Severity and Early Progression in Multiple Sclerosis. BMC Med Gen. 2012 (in press).
51) Molini E, Serafini G, Altissimi G, Simoncelli C, Ricci G. Sudden idiopathic
hearing loss. Case reports in the course of ten years. Acta Otorhinolaryngol Ital. 1998 Aug;18(4):218-27.