SORDITA’, AUTOSOMICA DOMINANTE 9; Gene COCH, DFNA9
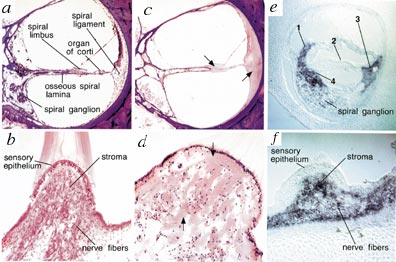
Fig1
Rapporti
fenotipo-Gene
|
Posizione |
Fenotipo |
Fenotipo |
Eredità |
Fenotipo |
Gene |
Gene |
|
Sordità, |
ANNO DOMINI |
COCH |

Diagram created by Andrew GNF based on data from Su AI,
Wiltshire T, Batalov S, et al (2004
TESTO
Un
simbolo di cancelletto (#) e ‘di questa scheda l’evidenza che DFNA9 è causata
da mutazione nel gene Cochlin (COCH; 603.196).
Qual è il nome
ufficiale del gene COCH?
Il nome ufficiale
di questo gene è “Cochlin.”
COCH è il
simbolo ufficiale del gene. Il COCH gene è conosciuto
anche con altri nomi, elencati di seguito.
Qual è la
funzione normale del gene COCH ?
Il COCH gene
fornisce istruzioni per fare una proteina chiamata Cochlin. Questa
proteina è abbondante in alcune parti dell’orecchio interno chiamato coclea e
sistema vestibolare. La coclea è una struttura a forma di chiocciola che trasforma
le vibrazioni sonore in impulsi elettrici ed il sistema vestibolare è
costituito da canali piene di liquido che servono per mantenere l’equilibrio
del corpo e l’orientamento nello spazio. Cochlin viene esportata dalle cellule
del sistema cocleare e vestibolare e diventa parte della matrice
extracellulare. La matrice extracellulare è un reticolo intricato che si
forma nello spazio tra le cellule e fornisce il supporto strutturale. Due
regioni della proteina Cochlin, chiamati domini LCCL e VWFA, probabilmente
coordinano le interazioni di Cochlin con altre molecole della matrice
extracellulare. Queste interazioni sono importanti nella formazione della
matrice extracellulare e mantenimento della organizzazione della matrice. Sebbene
il ruolo esatto Cochlin rimanga sconosciuta, probabilmente gioca un ruolo nella
struttura dell’orecchio interno.
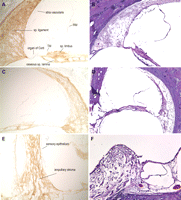
Figura
1. DFNA9 istopatologia ed espressione della COCH.
Microfotografia ![]() (50) di (un), il giro basale della
(50) di (un), il giro basale della
coclea da un membro inalterato di parenti 1W (59-anni-maschio, sconosciuto il tempo
post-mortem), e (b), il Cresta ampollare del canale
semicircolare posteriore da un osso controllo umano temporale (81-anno-maschio,
postmortem tempo 7 ore) colorata con ematossilina e eosina (H & E). I
componenti normali della coclea (a) e le normali costituenti
del crista (b sono mostrati). Microfotografie
(50) da due membri con problemi di udito di (c), il giro basale della
coclea (parenti 1St, 71 anni, di sesso maschile, il tempo di post-mortem
sconosciuto) e (d), cresta ampollare del canale semicircolare
posteriore ( parentado 1W, 86 anni, di sesso femminile, il tempo di post-mortem
18 h) colorate con H & E. C, La lamina spirale ossea, limbus spirale e
spirale legamento spettacolo perdita di cellularità con presenza di sostanza
acidofile coerente con-mucopolisaccaridi contenente sostanza fondamentale
(frecce) Vi è la perdita di cellule gangliari a spirale e dei loro
dendriti che innervano l’organo del Corti. Vi è anche la degenerazione
dell’organo del Corti. D, Lo stroma del cresta mostra sostanza
colorazione acidofile (frecce) simile a quello trovato nella coclea. C’è
perdita di cellule stromali e fibre nervose. L’epitelio sensoriale mostra
grave degenerazione delle cellule ciliate, con solo le cellule di supporto
rimanente. In situ ibridazione di posthatching giorno coclea
pollo (e) e ampollare crista (f) del
labirinto vestibolare con Coch antisenso ribosonda. Coch segnale
viene rilevato nello stesso . regioni come la sostanza acidofile trovato nella
coclea, e macule e creste del labirinto vestibolare dei pazienti
DFNA9 e, Coch messaggio è visto in cellule fusiformi lungo il percorso delle
fibre del nervo uditivo che penetrano nelle perforata habenula (4,
corrispondente alla lamina umana ossea spirale) tra i corpi cellulari gangliari
e dell’epitelio sensoriale, nella zona delle piastre cartilaginee dell’arto
neurale (1; corrispondente al limbus spirale umano) e l’arto abneural (3;
corrispondente al legamento spirale umana) sulla mediale e bordi laterali
dell’epitelio sensoriale. Nessun segnale è rilevato nella papilla basilare (2;
sensoriali dell’epitelio equivalente al organo umano di Corti), il tegmentum
vasculosum (equivalente al vascularis stria umana), o cellule gangliari
spirale. Crollo artefatti della vasculosum tegmentum (sopra l’epitelio
sensoriale) è visto. F,Coch segnale è rilevato nello stroma sottostante
l’epitelio sensoriale e la zona intorno alle fibre nervose vestibolari. Nessun
segnale viene rilevato nell’epitelio sensoriale (cellule ciliate e cellule di
sostegno).
Descrizione
DFNA9 è
una forma autosomica dominante ad insorgenza nell’età adulta di progressiva
ipoacusia neurosensoriale associata a disfunzione vestibolare variabili
(riassunto da Robertson et al., 2006). ![]()
Il gene COCH è probabilmente il gene più frequentemente coinvolto
in casi di sordità non sindromica ad eredità autosomica dominante (DFNA9). Si
tratta di una forma progressiva che inizialmente interessa soprattutto le alte
frequenze e che comporta anche disturbi dell’equilibrio (vertigini) a causa del
coinvolgimento di strutture dell’orecchio interno. La funzione del gene COCH
non è ancora del tutto nota, ma nell’orecchio interno delle persone affette
sono stati evidenziati depositi di lunghe molecole zuccherine (mucopolisaccaridi),
probabile causa della degenerazione delle fibre nervose.
Caratteristiche
cliniche
Manolis et al. (1996) riportarono
risultati di un’analisi genetica linkage in una famiglia con non sindromica
dell’udito neurosensoriale progressiva perdita postlinguale. In questa
perdita dell’udito famiglia è stata ereditata come carattere autosomico
dominante che inizia a circa 20 anni di età e progredisce alla sordità
totale. Manolis et al. (1996) descrissero
unici reperti istopatologici osso temporale in questa famiglia. Gli individui
affetti sono stati trovati ad avere deposizioni mucopolisaccaridi nei canali
del cocleare e nervi vestibolari. Queste deposizioni apparentemente
causato strangolamento e la degenerazione delle fibre dendritiche.Manolis et al. (1996) notarono
che altri (Khetarpal et al., 1991; Khetarpal, 1993)avevano
riferito precedenti valutazioni cliniche di questa famiglia. ![]()
Sulla base dei risultati delle 3 famiglie colpite, compresa la famiglia di Manolis et al.(1996), Robertson et al. (1998) ha
descritto la perdita dell’udito come avente il suo esordio tra i 20 ei 30 anni
di età. Inizialmente era più profonda alle alte frequenze e visualizzato
progressione variabile anacusis da 40 a 50 anni di età. Alcuni pazienti
DFNA9 avevano ricevuto gli impianti cocleari e altri utilizzati apparecchi
acustici.Uno spettro di clinica coinvolgimento vestibolare, che vanno dalla
mancanza di sintomi alla presenza di vertigine, ipofunzione vestibolare come
valutato dal elettronistagmografia e istopatologia, era stato trovato. ![]()
Come sono
cambiamenti nel gene COCH correlato alle condizioni di
salute?
Sordità
sindromica –
causata da mutazioni nel COCH gene
Diversi mutazioni
del gene COCH sono state identificate in individui con una forma
di sordità non sindromica (perdita di udito senza segni e sintomi correlati che
interessano altre parti del corpo) chiamato DFNA9. Queste sono mutazioni o
modificazioni o eliminazione di un blocco della costruzione di proteine
(aminoacidi) usate per fare Cochlin. La maggior parte dei mutazioni
del gene COCH segnalate influenzano il dominio LCCL e
probabilmente mettere in pericolo le interazioni di Cochlin con altre molecole
nella matrice extracellulare. Una mutazione avviene al di fuori del
dominio LCCL e probabilmente colpisce la forma dimensionale 3D di Cochlin.
Non è del
tutto chiaro come le mutazioni del gene COCH portano alla
perdita dell’udito e, in alcuni casi, a problemi di equilibrio e di orientamento. La
proteina Cochlin alterato potrebbe non riuscire a entrare a far parte della
matrice extracellulare causa di interazioni deteriorate con altre molecole, o
può interrompere l’organizzazione della matrice extracellulare. Inquietante
la matrice extracellulare può causare cambiamenti strutturali dell’orecchio
interno che influenzano l’udito e l’equilibrio. Cochlin sembra anche faccia
parte dei depositi anomali che si formano nell’orecchio interno delle persone
con sordità DFNA9. Questi depositi possono danneggiare i nervi che sono
fondamentali per la funzione uditiva.
Mappatura
Con
analisi del linkage in una famiglia con perdita dell’udito neurosensoriale
progressiva, postlinguale non sindromica, Manolis et al. (1996) hanno
dimostrato che la sordità è localizzata nel cromosoma 14q12-q13. Il massimo
punteggio lod (6.19 a teta = 0.0) è stato ottenuto con il marcatore
D14S121. ![]()
Dove
si trova il COCH trova gene?
Località
Citogenetica: 14q11.2-q13
Posizione
molecolare sul cromosoma 14: coppie di basi 30.874.535 a 30,895,079
(Homo sapiens Annotazione di uscita 107, GRCh38.p2) (NCBI)![]()
![]()

Il COCH gene
è situato sul ) braccio lungo (q del cromosoma 14 tra
le posizioni 13 e 11.2.
Più
precisamente, il COCH gene si trova dalla coppia di basi
30.874.535 di paia di basi 30.895.079 sul cromosoma 14.
Genetica
molecolare
Nella
famiglia originaria di Manolis et al. (1996) e
2 famiglie supplementari con DFNA9 identificati con le caratteristiche istopatologiche
di acidofile sostanza fondamentale nella coclea e vestibolare labirinto, Robertson et al. (1998) descritte
mutazioni nel gene separati COCH (603196,0 mila uno – 603.196,0003), che si esprime quasi esclusivamente nell’orecchio
interno. ![]()
Fransen et al. (1999) identificarono
una mutazione nel gene COCH (P51S,603196.0004) in 1 grande belga e 2 piccole famiglie olandesi con
autosomica dominante non sindromica progressiva perdita di udito neurosensoria
associata a disfunzione vestibolare. Superiore al 25% dei pazienti affetti
da questa mutazione hanno mostrato sintomi aggiuntivi, tra cui episodi di
vertigine, acufeni, la pienezza sonora, e la perdita di udito. Fransen et al. (1999) hanno
suggerito che il gene COCH può essere uno dei fattori genetici che
contribuiscono alla malattia Meniere(156000) e
che la possibilità di una mutazione COCH dovrebbe essere considerata in
pazienti con malattia di Meniere sintomi. ![]()
Usami et al. (2003) eseguito
analisi della mutazione COCH in una popolazione giapponese di 23 pazienti
appartenenti a famiglie indipendenti con autosomica dominante deficit uditivo,
4 dei quali hanno riportato sintomi vestibolari, e 20 pazienti affetti da
malattie Meniere. Usami et al. (2003) conclusero
che mutazioni nel gene COCH sono responsabili di una frazione significativa di
pazienti con autosomica perdita dell’udito ereditaria dominante accompagnata da
sintomi vestibolari, ma non per la perdita dell’udito dominante senza
disfunzioni vestibolari o malattia sporadica Meniere. Essi hanno
identificato una nuova mutazione puntiforme nel gene COCH (603196,0006) in un paziente con perdita di udito autosomica dominante e
sintomi vestibolari. ![]()
Via et al. (2005) eseguita
una scansione dell’intero genoma e analisi di linkage in un pedigree americano
con problemi di udito e perdita vestibolari e oculomotori disturbi. Un
punteggio lod coppie massima di 7,08 è stato ottenuto con l’indicatore
D14S1021, e una mutazione venne identificata in esone 12 del gene COCH(603196,0007) che cosegregate alla disfunzione uditiva. Via et al. (2005) stabilirono
che questa era la prima mutazione da segnalare al di fuori del dominio LCCL,
che è codificato da esoni 4 e 5. La perdita di udito e disfunzione vestibolare
era presente in un maschio di 17 anni, in questa famiglia, il più giovane hanno
riferito età di insorgenza di un membro della famiglia DFNA9.
Yuan et al. (2008) riportarono
di una grande famiglia cinese con DFNA9 confermata da analisi genetiche (603196,0008). Età all’insorgenza andava dalla seconda alla quinta decade
di vita, e c’era qualche evidenza di anticipazione genetica, anche se i
risultati possono essere stati causa di pregiudizio. I membri della
famiglia più colpiti (82%) hanno avuto tinnito al momento della comparsa di
perdita dell’udito.La perdita dell’udito prima colpito le alte frequenze e poi
coinvolto tutte le frequenze. Nel complesso, i pazienti hanno mostrato una
audiogramma contorno inclinata verso il basso. Anche se nessuno ha avuto
lamentele vestibolari clinici, studi dettagliati hanno mostrato evidenza di
difetti sottili. ![]()
Hildebrand et al. (2009) ha
riportato un 5-generazioni famiglia americana in cui i membri con non sindromica
sordità neurosensoriale e svalutazioni vestibolare, esclusi 2 pensiero per
rappresentare fenocopie sordità, ha avuto una mutazione nel gene P51S
COCH (603196,0004). Inoltre, 1 membro con la mutazione P51S aveva superiore
deiscenza del canale semicircolare bilaterale (SCCD). La famiglia era
legato a quelli riportati da {3,2: Fransen et al. (1999, 2001}), che
fornisce ulteriori prove di una mutazione fondatore. Hildebrand et al. (2009) raccomandata
TC ad alta risoluzione dell’osso temporale in pazienti con sordità DFNA9 legati
e screening per COCH in casi sporadici o familiari di canale superiore
deiscenza semicircolare. ![]()
In 3 pazienti non imparentati con SCCD e senza storia familiare della malattia
o della sordità, Crovetto et al. (2012) mutazioni
nel codificanti esoni e confini introne-esone del gene COCH esclusi. ![]()
Patogenesi
In mouse
e orecchio interno dell’uomo, Robertson et al. (2006) trovarono
che Cochlin immunostaining è stato limitato a tessuti di origine mesodermica; strutture
derivati neuroectodermiche chiaramente mancavano dell’espressione
Cochlin. Robertson et al. (2006) trovarono
che ossa temporali da pazienti con DFNA9 hanno mostrato grandi quantità di
depositi acellulare eosinofili Cochlin-immunoreattive contenuti in tutto il
legamento spirale, limbus, e lamina spirale ossea. Mice Coch-nulli non hanno
mostrato tale materiale, suggerendo che le mutazioni DFNA9 associate producono
un effetto dominante negativo. Robertson et al. (2006)suggeriva
che l’ostruzione di questi canali in DFNA9 si traduce in un danno neuronale
secondario e nella perdita dell’udito. ![]()
Sordità congenita
Circa il 25% degli individui
congenitamente sordi hanno una malformazione riconoscibile della capsula otica
[37]. Queste anomalie vanno da
difetti minori all’ aplasia totale della coclea. Queste malformazioni
creano sfide particolari per successo dell’impianto , tra cui le comunicazioni
ampiamente brevetti tra lo spazio fluido spinale e la scala perilinfatica
tramite un zona cribrosa malformata , causando
potenzialmente una perdita di liquido cerebrospinale; giustapposizione
anormale del vestibolare apparecchio
a coclea, aumentando la possibilità di stimolazione involontaria dei neuroni
vestibolari da un impianto cocleare; difficile un un accesso per
l’impianto cocleare per assenza della finestra rotonda e / o mal nervo facciale
posizionati, e cellule gangliari spirale anomale e diminuite situate in un
modiolo rudimentale e nel canale di Rosenthal (fig. 10).
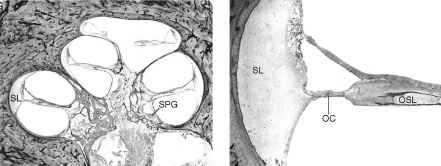
Fig. 9.
Questo – donna 59 anni ha subito un ipoacusia progressiva bilaterale
neurosensoriale a partire dall’età di 21 anni, secondaria ad una malattia
autosomica dominante (DFNA-9). Un audiogramma all’età di 50 anni ha
dimostrato una perdita di udito
neurosensoriale da grave a profonda in questo orecchio destro, con il 0% di
discriminazione vocale. Studio patologico dimostrato materiale
extracellulare eosinofila infiltrante il legamento spirale (SL) e la lamina
spirale ossea (OSL). Anche se c’erano residue cellule gangliari a spirale
(SPG) nel canale di Rosenthal, c’era l’atrofia totale dei dendriti periferici
della OSL. Cellule ciliate dell’organo del Corti (OC) erano presenti in
tutti e tre i giri della coclea. una sezione Midmodiolar. b potere
superiore di giro basale (line up b con a).
In sintesi, se non in
rari casi, l’istopatologia di grave sordità profonda nell’essere umano si trova
soprattutto nella parte interna dell’orecchio. È raro che vi sia totale
degenerazione delle cellule gangliari spirale, anche se la distribuzione e il
numero totale di queste cellule variano ampiamente.
Istopatologici
Cambiamenti nella coclea indotti da Cochlear Impianto
L’inserimento di un
array di elettrodi impianto cocleare provoca una quantità variabile di trauma
immediato e comporta anche effetti ritardati.
Trauma immediata causato da impianti cocleari
A seconda della posizione e le dimensioni della cocleostomia,
trauma significativo può verificarsi a membrana basilare e ossea lamina spirale
(figg. 11, 12).Spostamento della membrana basilare o frattura-lussazione della
spirale ossea
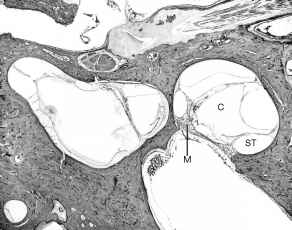
Fico. 10. Questo
85-year-old donna soffriva sordità congenita secondaria a grave displasia ossea
della coclea e sistema vestibolare. C’erano circa una metà
di giri nella coclea rudimentale (C). Non c’erano cellule ciliate. Il
modiolus (M) era rudimentale. Ci sono stati, tuttavia, alcune cellule
gangliari spirale rimanenti. La scala timpanica (ST) del giro basale è
stata marcatamente ridotta nelle dimensioni.
Fico. 10. Questo
85-year-old donna soffriva sordità congenita secondaria a grave displasia ossea
della coclea e sistema vestibolare. C’erano circa una metà di giri nella
coclea rudimentale (C). Non c’erano cellule ciliate. Il modiolus (M)
era rudimentale. Ci sono stati, tuttavia, alcune cellule gangliari spirale
rimanenti. La scala timpanica (ST) del giro basale è stata marcatamente
ridotta nelle dimensioni.
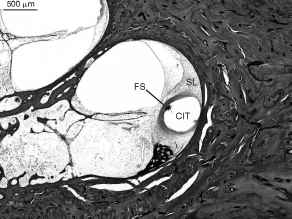
Fico. 11. Esempio di
trauma acuto causato da impianto cocleare in un uomo di 91 anni affetto da
sordità neurosensoriale progressiva e profonda bilaterale sottoposti impianto cocleare
dell’orecchio sinistro 11 anni prima della morte. L’impianto cocleare
pista (CIT) è visibile. In questo giro basale, la matrice dell’impianto
era penetrato la spirale legamento (SL) ed è stato circondato da una guaina
fibrosa (FS).
Fico. 11. Esempio di
trauma acuto causato da impianto cocleare in un uomo di 91 anni affetto da
sordità neurosensoriale progressiva e profonda bilaterale sottoposti impianto
cocleare dell’orecchio sinistro 11 anni prima della morte. L’impianto
cocleare pista (CIT) è visibile. In questo giro basale, la matrice
dell’impianto era penetrato la spirale legamento (SL) ed è stato circondato da
una guaina fibrosa (FS).
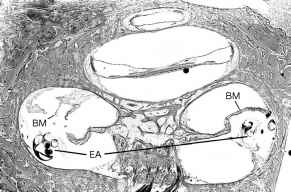
Fico. 12. trauma acuto
da impianto cocleare in una donna di 62 anni che ha subito una progressiva
perdita di udito e sordità profonda bilaterale come conseguenza di
aminoglicosidi ototossicità 5 anni prima dell’impianto. Nella sua
impiantati orecchio destro, la matrice di elettrodi (EA) è stata mantenuta in
situ.C’è frattura-lussazione della membrana basilare (BM) sulla sinistra a
circa 11 mm dalla membrana finestra rotonda, e lo spostamento della
membrana basilare (BM) a destra, a circa 18 mm dalla finestra rotonde membrana
dalla matrice di elettrodi.
Fico. 12. trauma acuto
da impianto cocleare in una donna di 62 anni che ha subito una progressiva
perdita di udito e sordità profonda bilaterale come conseguenza di
aminoglicosidi ototossicità 5 anni prima dell’impianto. Nella sua
impiantati orecchio destro, la matrice di elettrodi (EA) è stata mantenuta in
situ.C’è frattura-lussazione della membrana basilare (BM) sulla sinistra a
circa 11 mm dalla membrana finestra rotonda, e lo spostamento della membrana
basilare (BM) a destra, a circa 18 mm dalla finestra rotonde membrana dalla
matrice di elettrodi.
lamina non è raro
[38; vedi anche Roland e Wright, questo volume, pp 11-30]. Di tanto
in tanto, una rottura della membrana basilare può verificare con il passaggio
di un impianto cocleare da scala timpani in vestibuli Scala.
Danni al muro cocleare
laterali, in particolare nel tratto ascendente del giro basale, può
verificarsi.
Modello
animale
Makishima et al. (2005) ha
rilevato che Coch – / – mice senza Cochlin rilevabile nell’orecchio interno
avevano avuto risposte uditiva del tronco encefalico per fare clic e stimoli
puri toni indistinguibili da quelli di tipo selvatico topo. Un test lacZ
giornalista ha rivelato l’espressione di mRNA in Coch nonsensory regioni
epiteliali e stromali della coclea e del labirinto vestibolare nei topi
mutanti. Makishima et al.(2005) conclusero
che DFNA9 non può essere causato da COCH haploinsufficiency ma da un effetto
dominante negativo o il guadagno-di-funzione nelle regioni nonsensory
dell’orecchio interno. ![]()
|
RIFERIMENTI |
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Cochlin
immunocolorazione dell’ orecchio interno depositi patologici e analisi
proteomica in DFNA9 sordità e disfunzione vestibolare
4.
Tetsuo
Ikezono 3,
5.
Bryan
Krastiņš 4,
6.
Hannie
Kremer 2,
7.
Sharon F.
Kuo 1,
11. Joseph B.
Nadol Jr 5,
12. David A.
Sarracino 4,
13. Wim IM
Verhagen 6 e
14. Cynthia C.
Morton 1, *
+Autore Affiliazioni
1. 1 Dipartimenti
di Patologia, Ostetricia, Ginecologia e Biologia della Riproduzione, Brigham
and Women Hospital, Harvard Medical School, Boston, MA, Stati Uniti d’America,
2. 2
Dipartimento di Otorinolaringoiatria, Radboud University Medical Center,
Nijmegen, Paesi Bassi,
3. 3 Dipartimento
di Otorinolaringoiatria, Nippon Medical School, Tokyo, Giappone,
4. 4 Harvard
Medical School-Partners Healthcare Center di Genetica e Genomica, Cambridge,
MA, Stati Uniti d’America,
5. 5 Dipartimento
di Otology e Laringologia, Massachusetts Eye and Ear Infirmary, Eaton-Peabody
Laboratorio, Harvard Medical School di Boston, MA, USA e
6. 6 Dipartimento
di Neurologia, Canisius Wilhelmina Hospital, Nijmegen, Paesi Bassi
1.
* A chi corrispondenza deve essere indirizzata a: Dipartimenti
di Ostetricia, Ginecologia e Patologia, Brigham and Women Hospital, Harvard
Medical School, 77 Avenue Louis Pasteur, NRB 160, Boston, MA 02115, Stati Uniti
d’America. Tel: +1 6175254535; Fax: +1 6175254533; E-mail: cmorton @ partners.org
- Ricevuto 25
novembre 2005. - Accettato 7
FEBBRAIO 2006.
Astratto
Sette
mutazioni missense e una delezione mutazione in-frame sono stati segnalati nel
gene co fattore agulation C h omologia (COCH), causando
l’insorgenza nell’età adulta, sordità neurosensoriale progressiva e disturbo
vestibolare al locus DFNA9. Prevalenza di mutazioni COCH in
tutto il mondo non è nota, in quanto non vi è alcuno sforzo screening
sistematico per i disturbi uditivi ad esordio tardivo; Tuttavia, ad oggi,
le mutazioniCOCH sono stati trovati in quattro continenti e la possibilità
di COCHgiocare un ruolo importante nel presbycusis e disturbi di
squilibrio è stato considerato. Cochlin (codificata dal COCH) inoltre
è stato indicato come uno dei principali antigeni bersaglio per autoimmune
ipoacusia neurosensoriale. In questo rapporto, vi presentiamo
l’istopatologia, immunoistochimica e analisi proteomica di tessuti
dell’orecchio interno da post mortem DFNA9 campioni ossei temporale di un
individuo da un grande olandese affini segregante la mutazione e adulti P51S
controlli non affetti umani, e wild-type (+ / + ) e Coch nullo
(- / -) topi knock-out.DFNA9 è una malattia dell’orecchio interno con un
istopatologia unico che mostra la perdita di cellularità e aggregazione di
abbondanti depositi eosinofili acellulare omogenee nel cocleare e labirinti
vestibolari, simili a aggregazione proteica in ben noti malattie
neurodegenerative. Con immunoistochimica su sezioni di osso temporale
DFNA9, abbiamo dimostrato colorazione Cochlin dei caratteristici depositi
cocleari e vestibolari, indicando aggregazione Cochlin nelle stesse strutture
in cui è normalmente espresso. Analisi proteomica identificato Cochlin
come la proteina più abbondante nel mouse e coclee umana. L’espressione di
alto livello e stabilità Cochlin nell’orecchio interno, anche in assenza e
grave atrofia dei fibrociti che normalmente esprimono COCH, sono
mostrati attraverso questi studi e chiarire ulteriormente gli eventi
pathobiologic verificano in DFNA9 conseguente perdita e disfunzione vestibolare
dell’udito .
Sezione precedenteSezione successiva
INTRODUZIONE
Un gran
numero di loci sono stati mappati per sindromica e non sindromica sordità
ereditaria, e le mutazioni del gene responsabili di queste patologie vengono
continuamente scoperti e caratterizzati (1). Il
chiarimento delle funzioni di questi geni e il loro ruolo nella nell’orecchio
interno e in patogenesi di udito e disturbi dell’equilibrio sono importanti
sforzi in corso.
La sordità
malattia autosomica dominante nel locus DFNA9 è stata descritta e gli aspetti
clinici ampiamente caratterizzato, mostrando ad insorgenza nell’età adulta,
progressiva sordità neurosensoriale e disfunzione vestibolare (2 – 9). Diverse
mutazioni missense nel COCH (coagulation fattore C
h omologia) gene sono stati trovati inizialmente in tre famiglie negli
Stati Uniti, e successivamente in famiglie nei Paesi Bassi, in Belgio e
Australia (Tabella 1 e
Fig. 1) (10 – 14) . Sono
stati riportati – Due casi simplex di un’altra mutazione missense e un in-frame
delezione nello stesso dominio di COCH [h omologia
(FCH) / l imulus fattore C, c ochlin, lung
proteina gestazionale (LCCL) dominio f attore C] in
Giappone e Ungheria rispettivamente (15, 16). Un
recente rapporto descrive la prima scoperta di una mutazione COCH al
di fuori del dominio / LCCL FCH nel fattore A-simile (VWFA) dominio di von
Willebrand in un grande gruppo di affini DFNA9 negli Stati (Uniti 9). La
prevalenza delle mutazioni COCH in tutto il mondo non è nota,
come i test genetici sistematica di perdita di udito in età adulta non è
attualmente eseguita.

Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
Figura 1. Rappresentazione schematica
della struttura di amminoacidi corrispondente di COCH umana,
che codifica per la proteina Cochlin, mostra un peptide di segnale previsto
(SP), seguito da un dominio inizialmente designato come FCH, noto anche come
dominio LCCL, seguito da un dominio intermedio (IVd1) e due domini fattore di
von Willebrand A-simile (vWFA1 e vWFA2) separati da un dominio intervenendo
(ivd2). Sei mutazioni missense familiari (cinque nel dominio FCH / LCCL e
uno nel dominio vWFA2) causando DFNA9 sordità e disturbo vestibolare sono
indicati da frecce. Le posizioni di tutti i residui di cisteina sono
indicati come ‘C’. Le tre isoforme conosciute di Cochlin sono
rappresentati da linee orizzontali corrispondenti al loro sequenza e designati
come p60 (full-length, escludendo la SP) e due isoforme più corte (p44 e p40,
entrambi privi del dominio FCH / LCCL).L’anticorpo Cochlin utilizzato in questo
studio è stato effettuato nei confronti di un piccolo peptide N-terminale del
dominio vWFA1 (residui amminoacidici 163-181), mostrata in figura.Questo
anticorpo riconosce tutte e tre le isoforme di Cochlin.
Visualizza
questa tabella:
Tabella 1.
Mutazioni Coch nella DFNA9
COCH è
stato isolato inizialmente da approcci sottrattivi organo-specifiche da una
libreria di cDNA cocleare feto umano ed è risultato essere espresso ad alti
livelli nell’orecchio interno di macchia settentrionale, tessutoibridazione
in situ e immunoistochimica (17 – 20). La
proteina secreta, Cochlin, è stata rilevata mediante analisi proteomica come la
proteina più abbondante nella parte interna dell’orecchio bovina (21). Analisi
istopatologiche di ossa temporali DFNA9 colpite nei tre originali famiglie
statunitensi hanno rivelato preziose informazioni sulle modifiche apportate in
questi endpoint orecchio interno (3, 22). Un
risultato sorprendente e unico in queste ossa temporali, che in realtà ha
permesso l’identificazione iniziale di alcune di queste famiglie come parentele
DFNA9, è la presenza di eosinofili omogenee depositi extracellulari nelle
stesse zone, come l’atrofia fibrocyte. Altre patologie neurodegenerative
ben caratterizzati con accumulo di proteine aberranti comprendono
la malattia di Alzheimer (proteina precursore β-amiloide) (23), malattia
di Huntington (huntingtina) (24 – 26) e la
malattia di Parkinson (α-sinucleina)(27). Tuttavia,
DFNA9 è l’unico noto disturbo dell’orecchio interno che mostra questo tipo di
aggregato constatazione patologico firma, mentre i risultati in altre patologie
dell’orecchio interno, come idrope endolinfatico, e la degenerazione delle
strutture, come l’epitelio sensoriale, le cellule gangliari, legamento a
spirale e vascularis stria, si osservano in una varietà di differenti
condizioni mostrano perdita dell’udito e disfunzione vestibolare.
I fibrociti
cocleari e vestibolari, che sono gravemente atrofizzati in DFNA9, sono cellule
che esprimono COCH, ei depositi acellulari omogenei si trovano
nelle stesse zone Cochlin immunostaining nel normale dell’orecchio
interno (19). Tuttavia,
prima di questo rapporto, che non è stato dimostrato che questa sostanza
eosinofila nel DFNA9 è il Cochlin anormale che ha precipitato e aggregata o se
sia un altro componente dell’orecchio interno, una proteina-Cochlin
interagiscono, o qualche altro effetto a valle mutazioni COCH.
Un recente
donazione post mortem di un osso temporale, da un individuo dai Paesi Bassi con
DFNA9 (COCH P51S mutazione) ha fornito la possibilità di
ottenere dati istopatologici aggiuntive per DFNA9. Utilizzando un
anticorpo al dominio VWFA di Cochlin, che rileva tutte le isoforme di
dimensioni conosciute di Cochlin (Fig. 1), abbiamo
intrapreso una caratterizzazione approfondita delle Cochlin immunostaining nel
cocleare e labirinti vestibolari. Presentiamo analisi proteomica delle
sezioni ossee DFNA9 colpite e inalterati umani adulti temporali, così come di
wild-type (+ / +) e Coch nullo (- / -) topo knock-out orecchio
interno. Questi studi hanno permesso esame del contenuto dei depositi
anomali visti in DFNA9 e fornite informazioni sul ruolo del Cochlin
nell’orecchio interno e il meccanismo di pathobiology sottostante DFNA9 da
mutazioni COCH.
Sezione precedenteSezione successiva
RISULTATI E DISCUSSIONE
Istopatologia di
DFNA9 ossa temporali con lo Cochlin mutazione P51S
Alterazioni
anatomopatologiche in osso temporale P51S DFNA9 (Fig. 2),sono
coerenti con quelli precedentemente riportati per altre famiglie DFNA9
designati 1W (V66G mutazione), 1SU (G88E mutazione) e 1 ° (W117R) (3, 22, 28), suggerendo
che la mutazione P51S provoca le stesse modifiche patologiche e agisce
attraverso il medesimo meccanismo delle altre mutazioni nel dominio FCH / LCCL
di COCH. Una marcata riduzione del numero di fibrociti viene
osservato in tutto il legamento a spirale e limbus del condotto cocleare e
negli organi vestibolari. Nelle stesse aree di perdita fibrocyte, e
atrofia è la-eosinofila colorazione extracellulare sostanza fondamentale
DFNA9-caratteristica. I depositi sono particolarmente importanti nelle
parti più mediale del legamento sottostante vascularis stria e nella zona di
inserimento del legamento nella membrana basilare. Materiale eosinofila è
anche presente nella lamina spirale ossea, insieme con la perdita di dendriti
in questi canali e nel modiolus.
Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
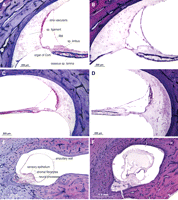
Figura 2. H & E macchiato sezioni
osso temporale celloidina-embedded da un individuo con DFNA9 da un olandese
gruppo di affini segregare la mutazione P51S (67 anni femmina) (B, D, F)
eda un controllo inalterato pari età (63 anni, femmina)(A, C, E). Nel
dotto cocleare dell’individuo DFNA9 colpite (B, D, × 100), in confronto con il
controllo inalterato (A, C, × 100), i risultati più sorprendenti sono la
presenza di abbondanti aggregati extracellulari eosinofile tutto il legamento a
spirale , spirale limbus e la lamina a spirale ossea, e la perdita
significativa e degenerazione fibrociti nel legamento e limbus. In
particolare, le parti più mediale del legamento a spirale, sottostanti
vascularis stria e la zona di inserimento del legamento nella membrana basilare,
risultano maggiormente colpite con presenza di depositi e atrofia fibrocyte. È
anche osservato degenerazione dell’organo di Corti e dei processi neurali
lamina spirale ossea. Nel ampolla del canale posteriore semicircolare nel
labirinto vestibolare (E, F, × 40) cambiamenti patologici, simili a quelli nel
condotto cocleare, sono presenti l’ampolla DFNA9 colpite (F) rispetto al
controllo inalterato (E). Abbondante deposizione eosinofila è presente
nello stroma ampollare DFNA9, con riduzione e atrofia dei fibrociti stromali,
nonché degenerazione dell’epitelio sensoriale del crista, e atrofia del nervo
ampollare. Inoltre, vi è ispessimento apprezzabile e parziale collasso
della parete ampollare, mostrando anche la presenza di aggregati eosinofili.
Cochlin
immunostaining in normale dell’orecchio interno
Prima di
eseguire immunoistochimica su ossa temporali DFNA9 colpite, abbiamo ottimizzato
anti-Cochlin colorazione anticorpale sulla normale mouse e tessuti umani
adulti, nonché su un Coch (- / -) topo (29, 30). Nei
nostri studi precedenti, abbiamo utilizzato un anticorpo policlonale per
l’intero FCH / LCCL e IVd1 dominio di Cochlin (19). Tuttavia,
date le isoforme diverse dimensioni di Cochlin rilevati da analisi proteomica e
sequenziamento N-terminale (21) (Fig. 1), quattro
anticorpi anti piccoli peptidi in diverse regioni del Cochlin sono stati
sviluppati e dimostrato da analisi Western Blot per essere specifici per il
isoforme che ogni era previsto di riconoscere (31). L’anticorpo
anti-Cochlin al dominio vWFA1 reagisce con tutte e tre le isoforme Cochlin
noti: p60 (full-length) e P44 e P40 (sia privo del dominio FCH / LCCL)
(Fig. 1). Abbiamo
scelto questo anticorpo (anti-Cochlin / dominio vWFA1) per i nostri studi,
perché avrebbe fornito una rappresentazione più completa della localizzazione
Cochlin nei tessuti non colpiti e interessati.
Nel normale
coclea topo adulto (Fig. 3), Cochlin
immunocolorazione è forte nel legamento a spirale e limbus spirale. Nel
legamento, la colorazione è più scuro nella cresta basilare, nei pressi della
membrana basilare e più debole della prominenza spirale. Cellule che
rivestono canale di Rosenthal ei canali della lamina spirale ossea sono
Cochlin-positivi, mentre i corpi cellulari gangliari cocleari ei processi
neurali sono Cochlin-negativi. Distinto Cochlin colorazione dei periciti
che circondano i vasi sanguigni nel modiolus e in tutto il dotto cocleare si
osserva. Al contrario, le aree adiacenti di tessuti circostanti ossei
mancano chiaramente Cochlin colorazione. Strutture Cochlin-negativi nel
condotto cocleare sono l’organo del Corti, tra cui l’epitelio sensoriale e
membrana tettoria, stria vascularis, membrana di Reissner, cellule gangliari
cocleari e loro processi neuronali. Nel labirinto vestibolare, creste
(Fig. 3 G)
mostra colorazione intensa Cochlin nei fibrociti e stroma sottostante
dell’epitelio sensoriale e nella parete ampollare. L’epitelio sensoriale,
i processi neuronali nello stroma ampollare e l’osso circostante e tessuti
connettivi sono tutti Cochlin-negativi.
Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
Figura 3.L’immunoistochimica su postnatale (5
mesi) (+ / +)(A, C, G) e Coch (-
/ -) (E)del mouse sezioni dell’orecchio interno con
anti-Cochlin.Immunocolorazione (sui pannelli a sinistra) appare come un
prodotto di reazione DAB bruno-rossastro; no di contrasto è stata
applicata su queste sezioni. Sezioni seriali colorate con H & E sono
mostrati in parallelo sui pannelli di destra (B, D, F, H). Nella
(+ / +) dotto cocleare (A, × 100; C, × 150), prominente Cochlin
immunocolorazione è presente in fibrociti e ECM per tutto il legamento a
spirale e limbus spirale.Cellule di rivestimento del canale di Rosenthal (che
circonda il ganglio spirale) ed i canali della lamina spirale ossea contengono
anche Cochlin, mentre i corpi cellulari gangliari cocleari ei processi neurali
sono negativi per Cochlin immunocolorazione.Distinti anelli perivascolari
intorno vasi sanguigni nel modiolus e in tutto il dotto cocleare sono
macchiati. Al contrario, le aree adiacenti di tessuti circostanti ossei
mancano chiaramente Cochlin colorazione. Le strutture della coclea che
mostrano l’assenza di espressione Cochlin sono l’organo del Corti, tra cui
l’epitelio sensoriale e membrana tettoria (TM), stria vascularis, membrana di
Reissner e cellule gangliari a spirale. Il condotto cocleare nel Coch (-
/ -) mouse (E, × 100) è stato utilizzato come controllo negativo e privo di
qualsiasi immunostaining Cochlin, confermando la specificità di questo
anticorpo. Nella (/ + +) crista dell’ampolla posteriori nel labirinto
vestibolare (G, × 200), intensa colorazione Cochlin si osserva nei fibrociti e
stroma sottostante dell’epitelio sensoriale, così come nella parete ampollare. Processi
neuronali nello stroma ampollare e l’osso circostante e tessuti connettivi
mancanza qualsiasi immunocolorazione. L’epitelio sensoriale è anche
completamente privo di qualsiasi colorazione Cochlin, come osservato nel
condotto cocleare.
Per
confermare la specificità anticorpale, ci immunostained sezioni da unCoch (-
/ -) topo (29, 30), e non
è stata rilevata la colorazione (Fig. 3 E). I
controlli negativi con il solo anticorpo secondario mostrano anche senza la
colorazione di fondo (dati non riportati). La colorazione intensa per
Cochlin nel (+ / +) del mouse orecchio interno corrobora la constatazione di
Cochlin mediante analisi proteomica come molto abbondante e stabile proteine
nella coclea e organi vestibolari.
Nel
controllo inalterato umano adulto orecchio interno (Fig. 4), viene
rilevato un modello simile di Cochlin colorazione. Cochlin
immunoreattività era prominente tutta la spirale legamento, limbus spirale e
all’interno della lamina spirale ossea. Immunocolorazione nel modiolus
stata osservata anche intorno ai vasi sanguigni (dati non mostrati). Come
nel topo, l’organo del Corti, i corpi cellulari neuronali e assoni centrali e
periferiche mancano di espressione Cochlin. I ossee e mesenchimali tessuti
esterni circostanti sono anche senza macchia. Nell’adulto umana labirinto
vestibolare, creste mostrano anche immunostaining nell’area dei fibrociti
stromali e una mancanza di colorazione nell’epitelio sensoriale sovrastante
adiacente. Il muro ampollare contiene anche Cochlin, come osservato in
sezioni del mouse.
Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
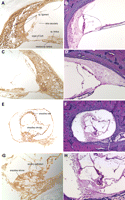
Figura 4.immunoistochimica su sezioni
dell’osso temporale con anti-Cochlin (A, C, E)inalterata
umano adulto (75 anni di sesso maschile). No di contrasto è stato
utilizzato su queste sezioni;sezioni H & E di serie sono riportati (B, D, F). Nella
dotto cocleare (A, C, × 100), Cochlin immunocolorazione è prominente in tutto
il legamento a spirale, limbus spirale ei canali della lamina a spirale ossea. Aree
adiacenti di tessuti circostanti ossei non sono macchiati con l’anticorpo
anti-Cochlin. Strutture della coclea mostrato in questa figura, che
mancano di espressione Cochlin, sono l’organo del Corti, tra cui l’epitelio
sensoriale e membrana tettoria (TM), stria vascularis e la membrana di Reissner
(RM). Alcune di queste strutture mostrano interruzione artefatta come
risultato di paraffina di ossa temporali adulti. Nei ampollare posteriori
crista del labirinto vestibolare (E, × 200), intenso Cochlin colorazione si
osserva nei fibrociti e stroma sottostanti dell’epitelio sensoriale. L’epitelio
sensoriale è completamente privo di qualsiasi espressione Cochlin, come è stato
osservato nel condotto cocleare.
In entrambi
gli organi del mouse e della coclea umana e vestibolari, Cochlin immunostaining
è limitato ai tessuti che sono mesodermal di origine; strutture
neuroectodermally derivati mancano chiaramente espressione
Cochlin. All’interno delle strutture mesodermiche, vi è diffuso e alto
livello di espressione di Cochlin in settori come il legamento a spirale, che
comprende una grande percentuale della massa totale della coclea membranosa, in
accordo con i risultati di elevati livelli di Cochlin mRNA di EST, Northern
blot e tessuti nelle analisi di ibridazione in situ(17, 19, 32), e con
abbondanza e stabilità di proteine Cochlin come osservato
mediante western blot e analisi proteomica (19, 21).
Cochlin
immunostaining in orecchio interno DFNA9 colpite
Il pattern
Cochlin colorazione nelle DFNA9 temporali sezioni ossee (Fig. 5)è simile a
quella in sezioni di controllo non affetti. L’immunocolorazione è forte in
tutto il legamento a spirale, a spirale limbus, stroma dei ampollare crista e
la parete ampollare. Come controllo negativo, è stata rilevata alcuna
colorazione con l’anticorpo secondario solo (dati non mostrati).Non c’è sfondo
rilevabile colorazione nei tessuti adiacenti alla parete laterale legamento
spirale e tessuti circostanti l’ampolla. Le regioni della lamina spirale
ossea normalmente occupata dalla assoni periferici cocleari sono immunopositive
per Cochlin, come pure le zone perivascolari nella modiolus. Altre
strutture come l’organo del Corti, vestibolari epitelio e stria vascularis
sensoriali, che sono Cochlin negativi nei tessuti normali, anche la mancanza di
colorazione.
Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
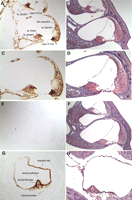
Figura 5.immunoistochimica su DFNA9 colpiti
umano adulto (67 anni femmina) sezioni dell’osso temporale con l’anticorpo
anti-Cochlin (A,C, E, G). No di contrasto è
stato utilizzato su queste sezioni; sezioni H & E di serie sono
riportati (B, D, F,H). Nella dotto
cocleare (A, C, × 100), Cochlin immunocolorazione è osservato in tutto il
legamento a spirale, limbus spirale ei canali della lamina a spirale ossea. I
depositi eosinofili omogenee visto sulle sezioni H & E sono macchiati scuro
e in modo uniforme. L’immunocolorazione Cochlin di questo materiale
acellulare è prominente nel legamento a spirale, in particolare nella zona di
inserimento nella membrana basilare, limbus a spirale e nei canali della lamina
a spirale ossea. L’organo del Corti e vascularis stria sono negativi per
Cochlin colorazione. Alcuni tessuti macchiati sottostante vascularis stria
sembra essere una parte del legamento spirale che è stato rimosso insieme al
stria. Aree adiacenti di tessuti ossei circostanti non mostrano alcun immunocolorazione. Nel
ampolla posteriore del labirinto vestibolare (E, × 4, G, × 100), Cochlin
colorazione dello stroma ampollare contenente i depositi eosinofili si osserva. Il
muro crollato ampollare mostrando ispessimento prominente e la deposizione acellulare
(F) contiene anche Cochlin (E). L’epitelio sensoriale non mostra
espressione Cochlin, come visto nel condotto cocleare sia DFNA9 e controllo
inalterato orecchio interno.
Le grandi
quantità di depositi acellulare eosinofili contenute in tutta la spirale
legamento, limbus e ossea spirale lamina sono scuro e in modo uniforme
immunostained con l’anti-Cochlin, ma completamente privi di colorazione non
specifica con solo anticorpo secondario. Lo stroma ampollare e la parete,
che mostrano la distorsione, il collasso e l’ispessimento, e contengono il
materiale acellulare, mostrano anche prominente Cochlin colorazione. Questi
risultati sono coerenti con l’idea che Cochlin è intimamente associata con i
depositi eosinofili caratteristici della istopatologia dell’osso temporale in
DFNA9.
Analisi proteomica
nel topo dell’orecchio interno
Analisi
proteomica del cocleare e labirinti di vestibolari (+ / +) e Coch (-
/ -) topi sono stati eseguiti e una lista ridotta del peptide rappresentante le
partite sono presentati (Tabella 2). Il numero
di peptidi triptici identificate da analisi di spettrometria di massa riflette
l’abbondanza relativa di proteine rilevate entro ciascun tessuto
con questo metodo. Una scoperta sorprendente è la presenza di Cochlin
peptidi come la proteina più abbondante rilevato nella coclea (+ / +) topi. Negli
organi vestibolari, Cochlin è la seconda proteina più frequentemente rilevata,
con l’albumina essere primario. In entrambi i tessuti, Cochlin è più
abbondante di β-emoglobina. I risultati per le altre due proteine,
α-tectorin e cheratina 9, sono rappresentativi di proteine
strutturali espresse in questi tessuti. La nostra analisi
proteomica combina i risultati di quattro frazioni gel dove la digestione e la
spettrometria di massa sono stati eseguiti separatamente.Identificazione
Cochlin come la proteina più abbondante nel topo lisati dell’orecchio interno
corrobora precedente analisi proteomica mediante il metodo alternativo di
elettroforesi su gel 2D, rivelando anche Cochlin come la proteina più
prevalente in nell’orecchio interno bovina (21). Come
controllo negativo, abbiamo studiato il Coch (- / -) del
mouse. Nessun peptidi Cochlin stati rilevati sia in tessuti cocleari o
vestibolari, confermando l’assenza di proteine Cochlin come
indicato anche dal nostro immunoistochimica e da precedenti occidentale
blot (30).
Visualizza
questa tabella:
Tabella 2.
Abbondanza relativa di peptidi dell’orecchio
interno in wild-type Coch + / + e – / – mice
In (+ / +)
topo coclea, un totale di 123 Cochlin peptidi triptici rappresentano 38 peptidi
unici vengono rilevati, che vanno da 7 a 35 residui amminoacidici ciascuno. La
copertura peptide esteso per Cochlin è stata osservata (Fig. 6) in
tutti i domini della proteina full-length (escluso il peptide segnale). In
concomitanza con rilevamento di Cochlin come altamente abbondante e stabile
proteine nella cocleare e organi vestibolari, e la sua
espressione piuttosto limitato ad un livello elevato nell’orecchio interno, è
interessante notare che diversi studi hanno implicato Cochlin come antigene
bersaglio per autoimmune ipoacusia neurosensoriale via sia immunoglobuline e
meccanismi delle cellule T-mediata. Sono stati rilevati livelli sierici di
immunoglobuline anti-Cochlin in un numero di pazienti con perdita dell’udito
autoimmune (33, 34).Inoltre,
Cochlin ha dimostrato di co-immunoprecipitato con colina transporter-like
protein 2 come bersagli di perdita dell’udito anticorpi indotta (35, 36). Gli
studi hanno anche dimostrato sperimentalmente indotta CD4 + T-cellule-mediata
perdita di udito autoimmune con Cochlin come antigene bersaglio (37, 38). Inoltre,
recenti indagini hanno rivelato frequenze significativamente più elevati di
circolanti cellule T specifiche per Cochlin così come elevati titoli
anticorpali siero specifico-Cochlin in individui con autoimmune perdita
dell’udito neurosensoriale, rispetto ai controlli di pari età non affetti (39). Queste
relazioni implicano Cochlin come un importante obiettivo orecchio interno
antigene sia anticorpi e cellule T-mediata perdita dell’udito autoimmune.

Visualizza
versione più grande:
·
Scarica
come diapositiva di PowerPoint
Figura 6. Amino sequenza di acido di
Cochlin umana e mouse, mostrando la distribuzione di digerire frammenti
triptici identificati mediante spettrometria di massa dal tessuto cocleare. I
domini di Cochlin sono sottolineati ed etichettati (FCH / LCCL in rosso, vWFA1
e vWFA2 in blu), così come la SP, spaccati in Cochlin maturo e quindi non
rilevato tra frammenti triptici. Residui di amminoacidi identici tra uomo
e topo sono indicati da un punto. La copertura peptide Cochlin, come si
trova nella nostra analisi proteomica, da sezioni umani affetti fissati in
formalina e inclusi in paraffina temporali osso è evidenziato in giallo e da
topo fresco intero tessuto cocleare è evidenziata in verde.
Analisi proteomica
di umano adulto ossa non affetti e DFNA9-colpite e temporali
Perché
l’unico materiale disponibile dai campioni umani DFNA9 colpiti e non colpiti è
fissato in formalina, ossa temporali inclusi in paraffina, abbiamo dovuto
adottare un approccio diverso rispetto a quello utilizzato nel topo. Così,
l’analisi proteomica è stata eseguita su proteine estratte dalle
sezioni in paraffina 8-micron, mentre sono tipicamente utilizzati lisati
tissutali intero freschi o tessuti congelati.
Nel campione
dell’osso temporale inalterato umano adulto, Cochlin è anche la proteina più
abbondante rilevato mediante spettrometria di massa, come è avvenuto nel topo
utilizzando lisati freschi cocleari. Un totale di 66 peptidi Cochlin,
rappresentando 17 peptidi unici, vengono rilevati (Tabella 3), che
vanno da 10 a 35 aminoacidi residui ciascuna.Peptidi Cochlin identificati nel
campione umano sono rappresentative tutta la proteina in tutti i domini di
Cochlin, anche se la copertura peptide non è completo come quello trovato nel
campione mouse (Fig. 6). Questo
non è sorprendente dato che l’estrazione del peptide da sezioni in paraffina
fissati in formalina di archiviazione (40) è
molto più difficile che da fresche lisati tissutali totali. Ciononostante,
questi risultati indicano che Cochlin è anche un componente diffusa e stabile
dell’orecchio interno umano adulto.
Visualizza
questa tabella:
Tabella 3.
Proteine identificate da
DFNA9-affetti e non affetti adulti ossa temporali umane
Un elenco
completo alfabetico di tutte le proteine rilevate dall’analisi
proteomica delle sezioni dell’osso temporale umano adulto è presentato nella
Tabella 3. Le
principali classi di proteine trovate sono componenti
extracellulari e strutturali della coclea, Cochlin essendo il primario. Molte
delle proteine sono noti per essere espresso nel legamento
spirale, che comprende una quantità preponderante della massa totale cocleare. Tipi
di collagene I, II, IX e XI, che sono anche in abbondanza rilevati nella nostra
analisi, sono molto rappresentative delle componenti noti e stabili di tessuto
cocleare e coinvolti in entrambi i tipi non sindromiche e sindromiche di
perdita dell’udito (1). Altre
proteine spesso rappresentate nella nostra analisi sono quelli
che comprende elementi del citoscheletro, come cheratina, β-actina e
tubulina, che sono noti anche per essere espresso nella coclea. Un totale
di 13 peptidi unici sono identificati, che rappresentano cinque cheratine
distinte. Il filamento intermedio, vimentina, è la seconda proteina più
prevalente rilevato nella nostra analisi (29 partite peptidici, riflettendo 17
peptidi unici), seguito da collagene di tipo II, l’alfa 1 (11 partite
peptidici, che rappresentano due peptidi unici).Localizzazione
immunoistochimica di vimentina nell’orecchio interno gerbillo mostra
l’espressione ad alto livello di questa proteina in maggior parte delle cellule
del tessuto connettivo e in vari tipi di cellule epiteliali, compresi alcuni
tipi di organi di cellule di supporto Corti (41).
La nostra
analisi proteomica di sezioni di tessuto fissate in formalina e inclusi in
paraffina adulti umani ha alcune limitazioni nel numero e la complessità delle
proteine cocleari identificato, probabilmente a causa della
inaccessibilità e la possibile degradazione delle proteine
durante il processo di estrazione di quantità molto piccole di
tessuto in sezioni di paraffina di 8 micron. Tuttavia, anche con queste
limitazioni, sono stati rilevati molte proteine rappresentative. Cochlin
è la più diffusa, mostrando oltre il doppio dei peptidi rilevati come vimentina
e riflette la sua alta espressione e la stabilità anche dopo il protocollo di
trattamento e l’estrazione dai tessuti fissi ed embedded.
Nell’analisi
proteomica delle sezioni dell’osso temporale DFNA9 colpite (Tabella 3), sono
sostanzialmente meno proteine identificate, e anche meno
corrispondenze peptide per ogni proteina. Tuttavia, vi è una significativa
sovrapposizione nelle proteine che sono stati identificati nei
campioni DFNA9 colpiti e non colpiti. Oltre a componenti del sangue,
l’albumina e a- e beta-globine, principali proteine che si
trovano nell’osso temporale DFNA9 colpiti (e anche presenti nel campione
inalterata) sono Cochlin, tipi di collagene I Alpha 1 e alfa 2, e cheratine 1 e
2a. Un confronto quantitativo di proteine attraverso il
DFNA9 colpite e campioni costanti non possono essere effettuate in quanto i due
campioni variavano notevolmente in complessità e il numero di peptide totale
partite per ogni proteina, probabilmente a causa della maggiore difficoltà di
estrazione e solubilità delle proteine nel sezioni DFNA9 che era
evidente durante il trattamento dei campioni (anche discusso sotto). Per
ogni proteina identificata nel campione DFNA9, il numero totale di peptide
corrisponde varia da 1 a 3, mentre nel campione inalterato stati trovati 1 a 66
corrispondenze. Ad esempio, nel campione inalterato, il numero totale di
partite peptidici sono 66 per Cochlin, cinque per la cheratina 1 e due per il
collagene di tipo I alfa 2; corrispondenti partite peptide nel campione
DFNA9 colpite sono tre, uno e due, rispettivamente. Il numero di peptidi
Cochlin trovato nel campione DFNA9 è superiore o pari a quella di qualsiasi
altra proteina. Peptidi Cochlin individuati nel campione DFNA9 sono sia
dal N- e le porzioni C-terminale del Cochlin spanning aminoacidi residui da 81
a 91, nel dominio e aminoacidi FCH / LCCL residui 526-539 nel dominio vWFA2.
Un fattore
importante nella identificazione di proteine è stata che la
composizione del tessuto di campioni ossei temporali DFNA9 colpite ha
presentato una sfida per l’estrazione di proteine totali a causa
della insolubilità relativa di questi campioni. Gran parte di questo
campione è rimasto come aggregati insolubili durante il protocollo di
estrazione.Pertanto, un gran numero di proteine sono stati
probabilmente non solubilizzato o estratta, rendendoli inaccessibili per
l’analisi di spettrometria di massa e conseguente minore complessità e del
numero di proteine totali, nonché un numero complessivo inferiore
di peptidi. La difficoltà di estrazione e la solubilità delle proteine
dalle sezioni DFNA9 colpiti forse non è sorprendente dato
l’osservazione delle grandi quantità di DFNA9 aggregati eosinofili nel
legamento spirale e limbus al microscopio ottico. Tuttavia, Cochlin viene
rilevato nel tessuto DFNA9 colpite almeno tutte le volte che altre proteine
importanti come collagene, cheratina e componenti del sangue
albumina e globine. Rilevamento Cochlin nell’osso temporale DFNA9 come una
delle proteine primarie identificate mediante analisi proteomica
suggerisce la stabilità Cochlin aggregati anche in assenza e grave
degenerazione dei fibrociti che normalmente esprimono COCH e
corrobora nostra scoperta immunoistochimica di intensa Cochlin colorazione
della depositi abbondanti presenti nell’orecchio interno DFNA9 colpite.
Analisi RT-PCR
Per valutare
se l’allele COCH mutante in individui con le P51S mutazione
(nucleotide C207T) mostra espressione stabile del COCH trascritto
mutante, inversa reazione a catena di trascrizione-polimerasi (RT-PCR) è stata
effettuata su RNA da virus di Epstein-Barr (EBV) – linee di cellule
trasformate. Una banda distinta della dimensione attesa è stato ottenuto
(dati non mostrati) utilizzando primer fiancheggianti la mutazione.Cromatografi
sequenza mostrano altezze approssimativamente uguali sia per i normali e mutati
coppie di basi nel campione DFNA9 colpite eterozigote e un unico picco di
ampiezza superiore per la coppia di basi normale nel campione inalterato.
Questi
risultati suggeriscono che il trascritto mutante COCH mostra
espressione stabile in pazienti P51S DFNA9 e non è sottoposto a degradazione. Dato
che questo mutazione missenso e tutte le altre mutazioni COCH noti
(sette missense e un in-frame delezione) non causano alcun scioglimento
anticipato o troncamento della sequenza proteica previsto, si può prevedere che
l’allele mutante avrebbe mostrato espressione stabile. Tuttavia, alcune
mutazioni missenso producono trascritti o proteine instabili, che
sono sottoposti alla degradazione da meccanismi cellulari e efficacemente
provocano mancanza di una proteina funzionale. Nel caso di DFNA9,
rilevamento stabile del trascritto mutanteCOCH è coerente con
l’ipotesi di un negativamente dominante effetto della mutazione in DFNA9
patologia piuttosto che haploinsufficiency di Cochlin.
Cochlin deposizione
in DFNA9
I nostri
studi precedenti hanno dimostrato Cochlin essere secreto e glicosilata in
cellule di mammifero trasfettate con full-length COCH cDNA(42). Come
glicoproteina extracellulare, con le / LCCL e VWFA domini FCH, Cochlin rischia
di legarsi a, o interagire con altri componenti cellulari come proteine
extracellulari, glicoproteine e proteoglicani. Tali
interazioni sono stati indicati per altre proteine contenenti
questi tipi di domini (43 – 47). I
nostri studi di microscopia luce delle sezioni DFNA9 hanno identificato il
materiale eosinofila prominente come un materiale omogeneo extracellulare
acellulare. Colorazione Movats pentachrome suggerisce che questo materiale
può contenere una sostanza mucopolysaccharide simile(3). Esame
al microscopio elettronico di sezioni DNFA9 orecchio interno mostra questi
depositi extracellulari di essere altamente ramificata, disordine, sostanza
microfibrillare, con granuli di glicosaminoglicani simili sparse (48). Queste
osservazioni sono coerenti con la nostra scoperta di Cochlin immunocolorazione
della sostanza fondamentale eosinofila, suggerendo che questi aggregati
contengono extracellulare depositati Cochlin. È anche possibile che altre
proteine associate con Cochlin possono essere presenti e che la
natura di queste interazioni è alterata a causa della presenza di Cochlin
mutato.
In termini
di meccanismo reale delle mutazioni missenso conducono al misfolding e
aggregazione di Cochlin, diversi studi in vitro sono stati
condotti per indagare questa possibilità. Gli studi iniziali del dominio
FCH / LCCL di Cochlin nelle cellule batteriche hanno mostrato misfolding di
questo dominio dovuti a diverse dei noti mutazioni che causano malattie e le
precipitazioni del mutante FCH / LCCL peptide durante il processo di piegatura (49). I
nostri trasfezioni transitorie di full-length COCH con molte
delle mutazioni ereditarie non hanno evidenziato differenze di secrezione o
apparenti livelli steady-state di Cochlin (9, 42). Un
altro studio confermato questi risultati e riportato differenze Cochlin
deposizione nella matrice extracellulare (ECM) delle cellule in coltura,
suggerendo alterata integrazione Cochlin mutato nella matrice (50).Tuttavia, un
avvertimento di studi in vitro è la mancanza dell’ambiente
extracellulare appropriata dell’orecchio interno. Inoltre, l’insorgenza
tardiva e progressività di DFNA9 suggerisce che gli effetti di mutanti Cochlin
come aggregazione o alterata interazione con altri componenti ECM potrebbe
essere un processo cumulativo. Un modello di topo DFNA9 sarebbe meglio
affrontare a lungo termine e cambiamenti progressivi nell’orecchio interno a
seguito di Coch mutazioni e tale modello è attualmente in
corso di valutazione nel nostro laboratorio.
Possibile eziologia
e la patogenesi della DFNA9
Cochlin
immunocolorazione di aggregati eosinofile in DFNA9 e la grave atrofia dei
fibrociti nelle stesse zone puntano verso i siti primari dove cambiamenti
patologici potrebbero hanno avviato a seguito di mutazioniCOCH. Altre
osservazioni nelle sezioni post mortem DFNA9 sono la degenerazione neuronale
neuroepiteliale e nella parte interna dell’orecchio. Poiché Cochlin non è
espresso in queste strutture neuroectodermici, è probabile che questi
cambiamenti sono secondari a quelli nei tessuti mesodermici. La riduzione
notevole e la degenerazione di fibrociti e la sostituzione da aggregati in
tutto il legamento a spirale e limbus spirale sono negli stessi siti come i
percorsi di K + riciclaggio da cellule epiteliali dell’organo del Corti nel
vano dei media Scala endolinfatico (51), indicando
una rottura dell’integrità della rete di giunzioni che esiste normalmente tra
queste cellule e svolge un ruolo critico nella omeostasi di ioni necessari per
il corretto funzionamento delle cellule dei capelli. Pertanto, una
perturbazione di orecchio interno equilibrio ionico probabile si verifica in
DFNA9 a causa della mancanza di fibrociti e presenza di depositi in tutte le
zone critiche per K + riciclaggio.
Un’altra
osservazione in entrambe le orecchie interne non interessate e DFNA9 è Cochlin
immunocolorazione tutta la lamina spirale ossea e nelle aree modiolar neuroni e
loro processi circostanti, ma non nei corpi o processi cellulari neuronali. La
rilevazione di depositi Cochlin all’interno di questi canali neurali suggerisce
che l’ostruzione di questi canali e danno neuronale può anche essere che si
verificano a seguito di aggregati mutanti Cochlin.
Una scoperta
intrigante è Immunostaining aree perivascolari nella modiolus e in tutto il
dotto cocleare. Tale constatazione suggerisce la possibilità che Cochlin
deposizione in aggregati perivascolari di organi al di fuori dell’orecchio
interno è implicato in alta prevalenza di disturbi vascolari che è stato
trovato in alcune parentele P51S DFNA9 (4), tra
cui quello a cui l’attuale 67 anni -vecchio individuale femminile apparteneva(52, 53). Queste
osservazioni giustificano ulteriori studi di espressione Cochlin dei vasi
sanguigni extralabyrinthine.
Un’altra
osservazione interessante è la mancanza di qualsiasi altro reperto
istopatologico simili tra le ossa DFNA9 colpite temporali e quelle di Coch(-
/ -) topo (a ~ 5 mesi di età) (30) (Fig 3 e 5). In
realtà, questi topi, che non esprimono Cochlin, non mostrano alcun apparenti
anomalie dell’orecchio interno e l’eventuale perdita dell’udito significativa,
anche se più tardi temporali punti devono ancora essere valutati. I nostri
studi mostrano Cochlin-colorazione depositi eosinofili in DFNA9, oltre alla
mancanza di patologia conclamata in Coch (- / -) topo a 5 mesi
di età, sono un ulteriore sostegno che questo disturbo non è probabilmente
dovuto al COCH haploinsufficiency, ma piuttosto un risultato
di effetti deleteri di un meccanismo molecolare di mutazioni missense COCH‘guadagno
di funzione’.
Depositi Cochlin
trovati nel glaucoma
Recenti
studi di proteomica hanno rivelato Cochlin come il più frequentemente rilevati
proteine mediante spettrometria di massa nella rete trabecolare
(TM) di occhi umani glaucomatosi, ma assente nelle normali occhi dei donatori
di controllo di pari età (54).L’immunoistochimica
ha rivelato depositi Cochlin-colorazione co-localizzazione con la sostanza
mucopolisaccaride nella TM intorno al canale di Schlemm negli occhi
glaucomatosi nell’umano e nel modello DBA / 2J il mouse per il glaucoma (54, 55). Analisi
Western blot ha mostrato un aumento dei livelli Cochlin con l’aumentare
dell’età in uomo e topo, con la progressione della malattia, così come una
diminuzione parallela collagene di tipo II, una componente importante della
normale TM. Si ipotizza che l’architettura alterato di questo tessuto può
causare ostruzione del flusso acquosa nell’occhio (54, 55). Questi
studi suggeriscono disregolazione di espressione Cochlin in un sottogruppo di
glaucomi umano e topo. Il ritrovamento di Cochlin depositi in questi occhi
è concomitante con un aumento della pressione intraoculare e precede danno del
nervo ottico e la degenerazione delle cellule gangliari (54, 55).Somiglianze
interessanti come Cochlin deposizione e danno neuronale esistere tra le
conclusioni di glaucoma e in DFNA9; studi paralleli potranno fornire una
visione in funzione Cochlin e il suo ruolo nei processi di malattia in questi
due sistemi sensoriali.
Sezione precedenteSezione successiva
MATERIALI E METODI
Fazzoletti
Tessuti di
topo sono stati ottenuti in base alle linee guida e protocolli approvati dal
Comitato di Harvard Scuola Medica permanente per animali (Boston, MA, USA). Ossa
temporali umani sono stati ottenuti in conformità con le linee guida stabilite
dai comitati ricerca umana al Massachusetts Eye e Ear Infirmary (Boston, MA,
USA) e il Canisio Wilhelmina Hospital (Nijmegen, Paesi Bassi).
Topi postnatale
(3-6 mesi di età) sono stati utilizzati in questo studio.Wild-type (+ / +) topi
sono stati utilizzati per la valutazione di istologia normale, localizzazione
Cochlin e analisi proteomica Coch. (- / -) Topo, utilizzato
come controlli negativi, erano un regalo di Drs Colin Stewart e Clara
Rodriguez (29). I topi
sono stati perfusi intracardiaca e tessuti fissati in paraformaldeide al 4%. Dopo
la rimozione della staffa dalla finestra ovale e foratura del finestra rotonda,
4% paraformaldeide fissativo è stato perfuso dolcemente attraverso la coclea. Orecchio
interno sono stati immersi in fissativo per 2-4 h, seguito da decalcificazione
in 120 m Macido etilendiamminotetraacetico
(EDTA) per 1 settimana a temperatura ambiente, e inclusi in paraffina da
procedure istologiche standard. Sezioni seriali sono stati ottenuti a
spessore 5-8 micron e utilizzati per la colorazione con ematossilina e eosina
(H & E) e per immunoistochimica.Per l’analisi proteomica, tessuti cocleari
e vestibolari stati sezionati separatamente e trattati come descritto in
‘Analisi proteomica’.
Per tessuti
umani, ossa temporali da una femmina di 67 anni, che era un membro di una
grande DFNA9 affini nei Paesi Bassi segregare la Cochlin mutazione P51S (5, 11), sono
stati donati e trattati presso il Laboratorio Otopathology del Massachusetts
Eye and Ear Infermeria, Boston, MA.Tempo di post-mortem per i tessuti ottenendo
era 6 ore. In questo individuo DFNA9, l’insorgenza della sordità
neurosensoriale bilaterale e squilibrio si è verificato intorno all’età di 40 anni
con una progressione di sordità profonda per età 63, che è stata documentata da
valutazione audiologica. Sintomi vestibolari consisteva di squilibrio
andatura, l’instabilità nel buio e oscillopsia. Esami vestibolari rivelato
bilaterale ipofunzione vestibolare periferica. Per i controlli non colpiti
(senza storia di perdita di udito), ossa temporali ottenuti da due donatori,
una femmina di 63 anni e un maschio di 75 anni, sono stati utilizzati. Tempi
post-mortem per l’ottenimento di tessuti di controllo erano 8 e 15 ore,
rispettivamente. Ossa temporali sono stati fissati in formalina al 10%, di
calce in EDTA e ridotte utilizzando lamette per contenere solo la capsula otica
e dell’orecchio interno. Per ottenere la morfologia ottimale di sezioni,
una delle ossa temporali DFNA9 (lato sinistro) è stato incorporato in
celloidina, come è standard con ossa temporali trasformati per la luce studio
microscopico. Immunostaining è impegnativo nelle sezioni
celloidina-embedded, anche se non c’è l’integrità del tessuto molto meglio in
questo mezzo (Fig. 2); Pertanto,
per facilitare immunoistochimica, l’altro osso temporale (lato destro) è stato
incorporato in paraffina. Questo è il primo e unico dell’osso temporale da
un membro della famiglia DFNA9 che si è inclusi in paraffina ad oggi,
facilitando così le nostre analisi. Per i controlli non colpiti, le ossa
temporali dal 63-year-old donna sono stati incorporati in celloidina, e quelli
dal 75-anno-vecchio maschio in paraffina. I campioni sono stati sezionati
in serie con uno spessore di 20 micron per celloidina e 8 micron per paraffina,
e sezioni selezionate colorate con H & E. Sezioni di paraffina sono
stati trattati per immunoistochimica e analisi proteomica come descritto di
seguito.
Immunoistochimica
L’immunocolorazione
è stata eseguita usando un anticorpo anti-Cochlin generato contro un peptide
nel dominio vWFA1 di Cochlin (Fig. 1),corrispondente
ad amino acidi 163-181 di residui Cochlin umana, identici ai residui negli
murino e Cochlin bovina (31). Anti-siero
è stato purificato attraverso una colonna di proteina A sefarosio, seguita da
cromatografia peptide-affinità. Questo anticorpo (anti-Cochlin / dominio
vWFA1) riconosce tutte e tre le isoforme diverse dimensioni di Cochlin
(Fig. 1).
Immunoistochimica
è stata eseguita come descritto in precedenza (19), ad
eccezione delle modifiche come descritto di seguito, tra cui la mancanza di
fase antigene recupero. Sezioni in paraffina da postnatale (+ / +) eCoch (-
/ -) topo, il controllo inalterata e DFNA9 colpite umano adulto ossa temporali
sono stati incubati con anticorpi anti-Cochlin / anticorpo dominio vWFA1 notte
a temperatura ambiente, lavate e incubate con un IgG biotinilato secondario
anti-coniglio (Vector Labs, Burlingame, CA, USA). L’immunocolorazione è
stata visualizzata mediante incubazione con il reagente Vectastain ABC (Vector
Labs) seguita da 3,3′-diaminobenzidina (DAB). Le sezioni non sono state
contrastate.
Analisi proteomica
Per
l’analisi proteomica nel topo, cocleare membranosa e labirinti vestibolari
stati sezionati separatamente da circa 3 mesi (+ / +) e Coch (-
/ -) topo. Lisati proteici sono stati preparati mediante incubazione dei
tessuti in un tampone di lisi [2% sodio dodecil solfato (SDS), 100 m di
bicarbonato M ammonio, 10 m M ditiotreitolo (DTT), pH 8,5] a 90 ° C per 10 min, 37 ° C per
60 min, con successiva sonicazione in 0,2% SDS, e l’incubazione a 90 ° C per 10
min. Tre cicli di sonicazione e bollente sono stati eseguiti, seguita da
alchilazione con 30 m M iodoacetamide. La
reazione è stata spenta con 10 m M DTT e
campioni sono stati separati mediante elettroforesi su gel SDS in 8-16% gel di
poliacrilammide. Gel erano di dimensioni frazionata in quattro sezioni,
decolorato con due lavaggi di metanolo al 50% e 5% di acido acetico, seguito da
tre lavaggi alternati di bicarbonato di ammonio e acetonitrile.Fette di gel sono
stati essiccati e successivamente sospeso singolarmente in tripsina (5,5 mg /
ml a 50 m M bicarbonato di ammonio) prima
dell’incubazione a 37 ° C per 18 ore per la digestione delle proteine. I
peptidi sono stati estratti con due risciacqui di 50 m M bicarbonato di ammonio e due risciacqui di 50% acetonitrile
e 0,1% di acido formico. I campioni sono stati preparati per spettrometria
di massa mediante liofilizzazione e reidratazione in 5% acetonitrile e 0,1% di
acido formico.
Per
l’analisi proteomica dell’orecchio interno umano adulto, gli unici materiali
disponibili erano, tessuti inclusi in paraffina fissati in formalina.Sezioni di
8 micron di spessore sono stati utilizzati dallo stesso osso temporale DFNA9
colpite usati per immunoistochimica. Per un controllo dell’osso temporale
umano adulto, fissati in formalina, inclusi in paraffina, 8 micron sezioni
spesse sono stati utilizzati da un 85-anno-vecchia femmina con otosclerosi, ma
senza mostrare istopatologia orecchio interno e con le strutture dotto cocleare
normali tra cui il legamento a spirale e limbus spirale.
Estrazione
di proteine da sezioni incluse in paraffina umani è stata
effettuata aggiungendo eptano e incubando a temperatura ambiente per 60 min,
seguito dall’aggiunta di metanolo alle proteine pellet
estratti. Gli estratti proteici sono stati resolubilized e sonicate nel 2%
SDS, 100 m Mbicarbonato di ammonio, 10 m M DTT, pH 8,5. Riduzione delle proteine è
stato raggiunto da campioni di ebollizione a 90 ° C per 20 minuti, seguita da
incubazione a 37 ° C per 60 min e alchilazione in 30 m Miodoacetamide per 60 min a temperatura ambiente. Tripsina
digestione delle proteine e la preparazione per spettrometria di
massa sono state eseguite come descritto per i campioni del mouse.
Per la
spettrometria di massa, i campioni sono stati eseguiti su una workstation LCQ
DECA XP più Proteome X (Thermo Electron Corporation, San Jose, CA, USA). Identificazioni
Peptide sono state effettuate utilizzando Sequest attraverso il browser
Bioworks 3.1 (Thermo Electron Corporation).Ricerche nelle banche dati sono
state effettuate utilizzando i database NCBI RefSeqHuman e RefSeqMurine
utilizzando cisteine statici carbamidomethyl modificati e
differenziali ossidato metionine, seguiti da ulteriori ricerche utilizzando
modifiche differenziali.
Reazione a catena
della polimerasi di trascrizione inversa
RT-PCR è
stata eseguita con RNA totale isolato da linee cellulari EBV-trasformati da due
individui P51S DFNA9, utilizzando il RNAeasy midi-kit (Qiagen, Leusden, Paesi
Bassi). sintesi del cDNA è stata eseguita come descritto (56) e la
PCR è stata eseguita per 35 cicli in condizioni standard utilizzando i seguenti
primers fiancheggianti la mutazione: forward 5′-ACCAGAGGCTTGGACATCAG-3
‘nell’esone 4 e reverse 5′-TTTGAGACTGGATGCCATTG-3’ nell’esone 5 . I prodotti
amplificati erano gel isolato e sequenziato utilizzando un kit di reazione ABI
PRISM Big Dye Terminator Cycle Sequencing V2 Ready e l’apparato ABI 3730
sequenziamento del DNA (Applied Biosystems, Foster City, CA, USA).
Sezione precedenteSezione successiva
MATERIALE SUPPLEMENTARE
Il materiale
supplementare è disponibile presso HMG Online. La figura complementare
mostra immunoistochimica eseguita su umano adulto sezioni dell’osso temporale
con il solo anticorpo secondario, come controllo negativo; viene rilevata
alcuna colorazione.
Sezione precedenteSezione successiva
RINGRAZIAMENTI
Siamo
particolarmente grati agli individui e alle loro famiglie per la donazione di
ossa temporali e una descrizione più dettagliata della istopatologia P51S DFNA9
è in preparazione per la pubblicazione.Vorremmo ringraziare il Dott Roderick
Bronson e Li Zhang al Dana Farber / Harvard Cancer Center Roditore
Histopathology Core. Ringraziamo anche Drs Colin Stewart e Clara Rodriguez
per il loro dono della Coch (- / -) topo. Questo lavoro è stato sostenuto da NIH / NIDCD
concede R01-DC03402 (a CCM), R01-DC0188 e P30-05209 (a MCL), il NIDCD Nazionale
Osso temporale, udito e l’equilibrio Patologia Registro delle risorse e dal
signor Axel Eliasen, e la salute e Labor Sciences Research Grants in Giappone
(Ricerca sulle misure per le malattie intrattabili, e disturbi sensoriali e
comunicativi) (TI).
Questo
manoscritto è dedicato da Cynthia Morton alla memoria di Craig Philip Morton,
scomparso il 9 luglio 2005 di cancro al pancreas, e che alla sua morte donò le
sue ossa temporali al NIDCD Nazionale Osso temporale, udito e l’equilibrio
Patologia Registro Resource ( http://www.tbregistry.org/) dopo aver lottato con
la malattia di Meniere durante la sua vita troppo breve.
Conflitto di
interessi dichiarazione. Nessuno ha dichiarato.
- ©
L’Autore 2006. Pubblicato dalla Oxford University Press. Tutti i
diritti riservati. Per Autorizzazioni, inviare un’e-mail:
journals.permissions@oxfordjournals.org
Riferimenti
References
1.
↵
Van Camp, G. and Smith, R.J.H.
(2005) Hereditary hearing loss homepage. URL: http://www.webhost.ua.ac.be/hhh/.
2.
↵
Halpin, C., Khetarpal, U. and
McKenna, M. (1996) Autosomal dominant progressive sensorineural hearing loss in
a large North American Family. Am. J.
Audiol., 5, 105–111.
3.
↵
Khetarpal, U., Schuknecht,
H.F., Gacek, R.R. and Holmes, L.B. (1991) Autosomal dominant sensorineural
hearing loss: pedigrees, audiologic and temporal bone findings in two
kindreds. Arch. Otolaryngol. Head Neck Surg.,117, 1032–1042.
4.
↵
Bom, S.J.H., Kemperman, M.H.,
De Kok, Y.J.M., Huygen, P.L.M., Verhagen, W.I.M., Cremers, F.P.M. and Cremers,
C.W.R.J. (1999) Progressive cochleovestibular impairment caused by a point
mutation in the COCH gene at DFNA9. Laryngoscope, 109, 1525–1530.
5.
↵
Verhagen, W.I.M., Bom, S.J.H.,
Huygen, P.L.M., Fransen, E., Van Camp, G. and Cremers, C.W.R.J. (2000) Familial
progressive vestibulocochlear dysfunction caused by a COCH mutation
(DFNA9). Arch. Neurol., 57, 1045–1047.
6. Verstreken,
M., Declau, F., Wuyts, F.L., D’Haese, P., Van Camp, G., Fransen, E., Van den
Hauwe, L., Buyle, S., Smets, R.E., Feenstra, L. et al. (2001)
Hereditary otovestibular dysfunction and Meniere’s disease in a large Belgian
family is caused by a missense mutation in the COCH gene. Otol.
Neurotol., 22, 874–881.
7. Kemperman,
M.H., Bom, S.J.H., Lemaire, F.X., Verhagen, W.I.M., Huygen, P.L.M. and Cremers,
C.W.R.J. (2002) DFNA9/COCH and its phenotype. Adv.
Otorhinolaryngol., 61, 66–72.
8. Lemaire,
F.X., Feenstra, L., Huygen, P.L.M., Fransen, E., Devriendt, K., Van Camp, G., Vantrappen,
G. and Cremers, C.W.R.J. (2003) Progressive late-onset sensorineural hearing
loss and vestibular impairment with vertigo (DFNA9/COCH): longitudinal
analyses in a Belgian family. Otol. Neurotol., 24,743–748.
9.
↵
Street, V.A., Kallman, J.C.,
Robertson, N.G., Kuo, S.F., Morton, C.C. and Phillips, J.O. (2005) A novel
DFNA9 mutation in the vWFA2 domain of COCHalters a conserved
cysteine residue and intrachain disulfide bond formation, resulting in
progressive hearing loss and site-specific vestibular and central oculomotor
dysfunction. Am. J. Med. Genet. A, 139, 86–95.
10.
↵
Robertson, N.G., Lu, L., Heller, S.,
Merchant, S.N., Eavey, R.D., McKenna, M., Nadol, J.B., Jr, Miyamoto, R.T.,
Linthicum, F.H., Jr, Lubianca Neto, J.F. et al. (1998)
Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness
with vestibular dysfunction. Nat. Genet., 20, 299–303.
11.
↵
de Kok, Y.J.M., Bom, S.J.H., Brunt,
T.M., Kemperman, M.H., van Beusekom, E., van der Velde-Visser, S.D., Robertson,
N.G., Morton, C.C., Huygen, P.L.M., Verhagen, W.I.M. et al. (1999)
A Pro51Ser mutation in the COCH gene is associated with late
onset autosomal dominant progressive sensorineural hearing loss with vestibular
defects. Hum. Mol. Genet., 8, 361–366.
12.
↵
Fransen, E., Verstreken, M.,
Verhagen, W.I.M., Wuyts, F.L., Huygen, P.L.M., D’Haese, P., Robertson, N.G.,
Morton, C.C., McGuirt, W.T., Smith, R.J.H. et al. (1999)
High prevalence of symptoms of Meniere’s disease in three families with a
mutation in the COCH gene. Hum. Mol.
Genet., 8, 1425–1429.
13.
↵
Kamarinos, M., McGill, J.,
Lynch, M. and Dahl, H. (2001) Identification of a novel COCH mutation,
I109 N, highlights the similar clinical features observed in DFNA9
families. Hum. Mutat., 17, 351.
14.
↵
Kemperman, M.H., De Leenheer,
E.M.R., Huygen, P.L.M., van Duijnhoven, G., Morton, C.C., Robertson, N.G.,
Cremers, F.P.M., Kremer, H. and Cremers, C.W.R.J. (2005) Audiometric, vestibular,
and genetic aspects of a DFNA9 family with a G88E COCH mutation. Otol.
Neurotol., 26, 926–933.
15.
↵
Usami, S., Takahashi, K.,
Yuge, I., Ohtsuka, A., Namba, A., Abe, S., Fransen, E., Patthy, L., Otting, G.
and Van Camp, G. (2003) Mutations in the COCH gene are a
frequent cause of autosomal dominant progressive cochleo-vestibular
dysfunction, but not of Meniere’s disease. Eur. J. Hum.
Genet., 11, 744–748.
16.
↵
Nagy, I., Horvath, M.,
Trexler, M., Repassy, G. and Patthy, L. (2004) A novelCOCH mutation,
V104del, impairs folding of the LCCL domain of cochlin and causes progressive
hearing loss. J. Med. Genet., 41, e9.
17.
↵
Robertson, N.G., Khetarpal,
U., Gutiérrez-Espeleta, G.A., Bieber, F.R. and Morton, C.C. (1994) Isolation of
novel and known genes from a human fetal cochlear cDNA library using
subtractive hybridization and differential
screening.Genomics, 23, 42–50.
18. Robertson,
N.G., Skvorak, A.B., Yin, Y., Weremowicz, S., Johnson, K.R., Kovatch, K.A.,
Battey, J.F., Bieber, F.R. and Morton, C.C. (1997) Mapping and characterization
of a novel cochlear gene in human and in mouse: a positional candidate gene for
a deafness disorder, DFNA9. Genomics, 46, 345–354.
19.
↵
Robertson, N.G., Resendes,
B.L., Lin, J.S., Lee, C., Aster, J.C., Adams, J.C. and Morton, C.C. (2001)
Inner ear localization of mRNA and protein products ofCOCH, mutated in
the sensorineural deafness and vestibular disorder, DFNA9.Hum. Mol. Genet., 10, 2493–2500.
20.
↵
Ikezono, T., Shindo, S.,
Ishizaki, M., Li, L., Tomiyama, S., Takumida, M., Pawankar, R., Watanabe, A.,
Saito, A. and Yagi, T. (2005) Expression of cochlin in the vestibular organ of
rats. ORL J. Otorhinolaryngol. Relat. Spec., 67, 252–258.
21.
↵
Ikezono, T., Omori, A.,
Ichinose, S., Pawankar, R., Watanabe, A. and Yagi, T. (2001) Identification of
the protein product of the Coch gene (hereditary deafness
gene) as the major component of bovine inner ear protein. Biochim.
Biophys. Acta, 1535, 258–265.
22.
↵
Merchant, S.N., Linthicum,
F.H. and Nadol, J.B., Jr (2000) Histopathology of the inner ear in DFNA9. Adv.
Otorhinolaryngol., 56, 212–217.
23.
↵
Selkoe, D.J. (2004) Cell
biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s
diseases. Nat. Cell Biol., 6, 1054–1061.
24.
↵
Davies, S.W., Turmaine, M.,
Cozens, B.A., DiFiglia, M., Sharp, A.H., Ross, C.A., Scherzinger, E., Wanker,
E.E., Mangiarini, L. and Bates, G.P. (1997) Formation of neuronal intranuclear
inclusions underlies the neurological dysfunction in mice transgenic for the HD
mutation. Cell, 90, 537–548.
25. Zoghbi,
H.Y. and Orr, H.T. (2000) Glutamine repeats and neurodegeneration.Annu. Rev.
Neurosci., 23, 217–247.
26.
↵
Waelter, S., Boeddrich, A.,
Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H. and Wanker, E.E. (2001)
Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies
as a result of insufficient protein degradation.Mol. Biol. Cell, 12, 1393–1407.
27.
↵
Masliah, E., Rockenstein, E.,
Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A. and
Mucke, L. (2000) Dopaminergic loss and inclusion body formation in
alpha-synuclein mice: implications for neurodegenerative disorders. Science, 287, 1265–1269.
28.
↵
Khetarpal, U. (1993) Autosomal
dominant sensorineural hearing loss: further temporal bone findings. Arch.
Otolaryngol. Head Neck Surg., 119, 106–108.
29.
↵
Rodriguez, C.I., Cheng, J.G.,
Liu, L. and Stewart, C.L. (2004) Cochlin, a secreted von Willebrand factor type
a domain-containing factor, is regulated by leukemia inhibitory factor in the
uterus at the time of embryo
implantation.Endocrinology, 145, 1410–1418.
30.
↵
Makishima, T., Rodriguez,
C.I., Robertson, N.G., Morton, C.C., Stewart, C.L. and Griffith, A.J. (2005)
Targeted disruption of mouse Coch provides functional evidence
that DFNA9 hearing loss is not a COCH haploinsufficiency
disorder.Hum. Genet., 118, 29–34.
31.
↵
Ikezono, T., Shindo, S., Li,
L., Omori, A., Ichinose, S., Watanabe, A., Kobayashi, T., Pawankar, R. and
Yagi, T. (2004) Identification of a novel Cochlin isoform in the perilymph:
insights to Cochlin function and the pathogenesis of DFNA9. Biochem.
Biophys. Res. Commun., 314, 440–446.
32.
↵
Skvorak, A.B., Weng, Z., Yee,
A.J., Robertson, N.G. and Morton, C.C. (1999) Human cochlear expressed sequence
tags provide insight into cochlear gene expression and identify candidate genes
for deafness. Hum. Mol. Genet., 8, 439–452.
33.
↵
Boulassel, M.R., Deggouj, N.,
Tomasi, J.P. and Gersdorff, M. (2001) Inner ear autoantibodies and their
targets in patients with autoimmune inner ear diseases.Acta
Otolaryngol., 121, 28–34.
34.
↵
Boulassel, M.R., Tomasi, J.P.,
Deggouj, N. and Gersdorff, M. (2001) COCH5B2is a target antigen of
anti-inner ear antibodies in autoimmune inner ear diseases. Otol.
Neurotol., 22, 614–618.
35.
↵
Kommareddi, P.K., Nair, T.S.
and Carey, T.E. (2003) Cochlin is expressed in the supporting cells of the
organ of Corti and co-precipitates with CTL2. ARO abstracts 26:58. Presented at
the 25th midwinter research meeting of the Association for Research in
Otolaryngology, Daytona Beach, FL, February 23–27, 2003.
36.
↵
Nair, T.S., Kozma, K.E., Hoefling,
N.L., Kommareddi, P.K., Ueda, Y., Gong, T.W., Lomax, M.I., Lansford, C.D.,
Telian, S.A., Satar, B. et al. (2004)
Identification and characterization of choline transporter-like protein 2, an
inner ear glycoprotein of 68 and 72 kDa that is the target of
antibody-induced hearing loss. J.
Neurosci., 24, 1772–1779.
37.
↵
Solares, C.A., Edling, A.E.,
Johnson, J.M., Baek, M.J., Hirose, K., Hughes, G.B. and Tuohy, V.K. (2004)
Murine autoimmune hearing loss mediated by CD4+ T cells specific for inner ear
peptides. J. Clin. Invest., 113, 1210–1217.
38.
↵
Billings, P. (2004)
Experimental autoimmune hearing loss. J. Clin.
Invest.,113, 1114–1117.
39.
↵
Baek, M.J., Park, H.M., Johnson,
J.M., Altuntas, C.Z., Jaini, R., Thomas, D.M., Ball, E.J., Robertson, N.G.,
Morton, C.C., Hughes, G.B. et al. (2006)
Increased frequencies of cochlin-specific T cells in patients with autoimmune
sensorinerual hearing loss. J. Immunol., in press.
40.
↵
Palmer-Toy, D.E., Krastins,
B., Sarracino, D.A., Nadol, J.B., Jr and Merchant, S.N. (2005) Efficient method
for the proteomic analysis of fixed and embedded tissues. J. Proteome
Res., 4, 2404–2411.
41.
↵
Schulte, B.A. and Adams, J.C.
(1989) Immunohistochemical localization of vimentin in the gerbil inner
ear. J. Histochem. Cytochem., 37, 1787–1797.
42.
↵
Robertson, N.G., Hamaker,
S.A., Patriub, V., Aster, J.C. and Morton, C.C. (2003) Subcellular localisation,
secretion, and post-translational processing of normal cochlin, and of mutants
causing the sensorineural deafness and vestibular disorder, DFNA9. J. Med.
Genet., 40, 479–486.
43.
↵
Colombatti, A. and Bonaldo, P.
(1991) The superfamily of proteins with von Willebrand factor type A-like
domains: one theme common to components of extracellular matrix, hemostasis,
cellular adhesion, and defense mechanisms.Blood, 77, 2305–2315.
44. Colombatti,
A., Bonaldo, P. and Doliana, R. (1993) Type A modules: interacting domains
found in several non-fibrillar collagens and in other extracellular matrix
proteins. Matrix, 13, 297–306.
45. Lee,
J.O., Rieu, P., Arnaout, M.A. and Liddington, R. (1995) Crystal structure of
the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell, 80,631–638.
46. Delrieu,
I., Waller, C.C., Mota, M.M., Grainger, M., Langhorne, J. and Holder, A.A.
(2002) PSLAP, a protein with multiple adhesive motifs, is expressed inPlasmodium
falciparum gametocytes. Mol. Biochem.
Parasitol., 121, 11–20.
47.
↵
Whittaker, C.A. and Hynes,
R.O. (2002) Distribution and evolution of von Willebrand/integrin A domains:
widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol.
Cell, 13, 3369–3387.
48.
↵
Khetarpal, U. (2000) DFNA9 is
a progressive audiovestibular dysfunction with a microfibrillar deposit in the
inner ear. Laryngoscope, 110, 1379–1384.
49.
↵
Liepinsh, E., Trexler, M.,
Kaikkonen, A., Weigelt, J., Banyai, L., Patthy, L. and Otting, G. (2001) NMR
structure of the LCCL domain and implications for DFNA9 deafness
disorder. EMBO J., 20, 5347–5353.
50.
↵
Grabski, R., Szul, T., Sasaki,
T., Timpl, R., Mayne, R., Hicks, B. and Sztul, E. (2003) Mutations in COCH that
result in non-syndromic autosomal dominant deafness (DFNA9) affect matrix
deposition of cochlin. Hum. Genet., 113, 406–416.
51.
↵
Steel, K.P. (1999)
Perspectives: biomedicine. The benefits of
recycling.Science, 285, 1363–1364.
52.
↵
Verhagen, W.I.M., Huygen,
P.L.M., Theunissen, E.J.J.M. and Joosten, E.M.G. (1989) Hereditary
vestibulo-cochlear dysfunction and vascular disorders. J. Neurol.
Sci., 92, 55–63.
53.
↵
Verhagen, W.I.M., Bom, S.J.H.,
Fransen, E., Van Camp, G., Huygen, P.L.M., Theunissen, E.J.J.M. and Cremers,
C.W.R.J. (2001) Hereditary cochleovestibular dysfunction due to a COCH gene
mutation (DFNA9): a follow-up study of a family. Clin.
Otolaryngol. Allied Sci., 26, 477–483.
54.
↵
Bhattacharya, S.K., Rockwood, E.J.,
Smith, S.D., Bonilha, V.L., Crabb, J.S., Kuchtey, R.W., Robertson, N.G.,
Peachey, N.S., Morton, C.C. and Crabb, J.W. (2005) Proteomics reveal Cochlin
deposits associated with glaucomatous trabecular meshwork. J. Biol.
Chem., 280, 6080–6084.
55.
↵
Bhattacharya, S.K., Annangudi,
S.P., Salomon, R.G., Kuchtey, R.W., Peachey, N.S. and Crabb, J.W. (2005)
Cochlin deposits in the trabecular meshwork of the glaucomatous DBA/2J
mouse. Exp. Eye Res., 80, 741–744.
56.
↵
Luijendijk, M.W., van de Pol,
T.J., van Duijnhoven, G., den Hollander, A.I., ten Caat, J., van Limpt, V.,
Brunner, H.G., Kremer, H. and Cremers, F.P. (2003) Cloning, characterization,
and mRNA expression analysis of novel human fetal cochlear cDNAs. Genomics, 82, 480–490.
Articles citing this article
- Sensory substitution in bilateral vestibular a-reflexic
patientsPHY2 (2015) 3 (5): e12385
- Detailed Hearing and Vestibular Profiles in the Patients
with COCH MutationsAnn Otol Rhinol
Laryngol (2015) 124 (1_suppl): 100S-110S
- Gene Expression
Profiles of the Cochlea and Vestibular Endorgans: Localization and
Function of Genes Causing DeafnessAnn Otol Rhinol
Laryngol (2015) 124 (1_suppl): 6S-48S
- Phenotype Analysis of an Australian DFNA9 Family with the
I109N COCH MutationAnn Otol Rhinol
Laryngol (2011) 120 (6): 414-421
- Proteomics, bioinformatics and targeted gene expression
analysis reveals up-regulation of cochlin and identifies other potential
biomarkers in the mouse model for deafness in usher syndrome type 1FHum Mol
Genet (2010) 19 (8): 1515-1527
- COCH Transgene Expression in Cultured Human Trabecular
Meshwork Cells and Its Effect on Outflow Facility in Monkey Organ Cultured
Anterior SegmentsIOVS (2010) 51 (4): 2060-2066
- Proteome bioprofiles distinguish between M1 priming and
activation states in human macrophagesJ. Leukoc.
Biol. (2010) 87 (4): 655-662
- Cochlin Expression in Anterior Segment Organ Culture Models
after TGF{beta}2 TreatmentIOVS (2009) 50 (2): 551-559
- A targeted Coch missense mutation: a knock-in mouse model
for DFNA9 late-onset hearing loss and vestibular dysfunctionHum Mol
Genet (2008) 17 (21): 3426-3434
- Phenotype Description of a Novel DFNA9/COCH Mutation, I109TAnn Otol
Rhinol Laryngol (2007) 116 (5): 349-357