IPOACUSIA da MUTAZIONI del DNA MITOCONDRIALE
ASSOCIATE a FORME SINDROMICHE e NON
LA CLASSIFICAZIONE DELLE MALATTIE MITOCONDRIALI
FORME SINDROMICHE
KSS (Kearns–Sayre Syndrome)
MERRF (Myoclonic Epilepsy and Ragged Red Fibers)
MELAS (Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes)
MIDD (Maternally Inherited Diabetes mellitus and Deafness)
LA CLASSIFICAZIONE DELLE MALATTIE MITOCONDRIALI esteso
MALATTIE MITOCONDRIALI NON SINDROMICHE
LA CLASSIFICAZIONE DELLE MALATTIE MITOCONDRIALI
L’identificazione di mutazioni del mtDNA ha fornito le basi per l’attuale classificazione dei disordini mitocondriali.
Un primo gruppo di malattie è caratterizzato dalla presenza di mutazioni del mtDNA, ad insorgenza sporadica, o a trasmissione materna. Un secondo gruppo è causato da mutazioni in geni nucleari che fanno parte o controllano la fosforilazione ossidativa (OXPHOS). Queste malattie sono spesso classificate sulla base delle sole alterazioni biochimiche rilevate dall’analisi dei tessuti affetti (soprattutto muscolo scheletrico), perché i geni responsabili ancora non si conoscono, anche se molti progressi sono stati recentemente compiuti in questo campo.
1. Mutazioni del mtDNA
A seconda delle caratteristiche molecolari e genetiche delle mutazioni del mtDNA, questo gruppo di difetti comprende sindromi dovute a riarrangiamenti** su larga scala del mtDNA, o a mutazioni puntiformi* del mtDNA.
Tab.I Malattie da mutazioni di geni mitocondriali (mtDNA)*
|
Riarrangiamenti del DNA mitocondriale (delezioni)** |
|
Sindrome di Kearns-Sayre (KSS) |
|
Oftalmoplegia Esterna Progressiva (PEO) |
|
Sindrome di Pearson (anemia sideroblastica e malassorbimento connatali) |
|
Mutazioni puntiformi* |
|
Neuropatia ottica ereditaria di Leber (LHON) |
|
Sindrome di NARP (neuropatia, atassia, retinite pigmentosa) |
|
Sindrome MILS (Sindrome di Leigh ereditata per via matrilineare) |
|
Encefalopatia mitocondriale con acidosi lattica e strokes (MELAS) |
|
Mioclono-epilessia con fibre “ragged-red”(MERRF) |
|
Miopatia e cardiomiopatia (MIMYCA) |
* eredità matrilineare
** forme quasi sempre sporadiche
Alterazioni qualitative del mtDNA
Si può trattare di delezioni parziali del mtDNA o, più raramente, di duplicazioni parziali. Entrambi i tipi di mutazione sono eteroplasmici, dato che coesistono sempre con una quota di mtDNA normale. Queste alterazioni grossolane del mtDNA sono quasi invariabilmente associate con tre principali presentazioni cliniche: la sindrome di Kearns-Sayre, l’Oftalmoplegia Esterna Progressiva, e la sindrome di Pearson.
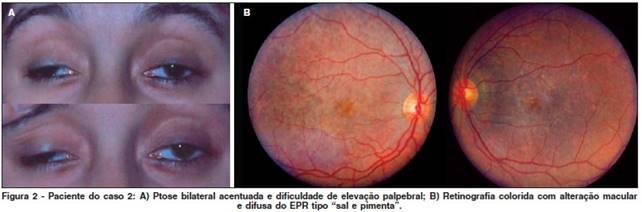
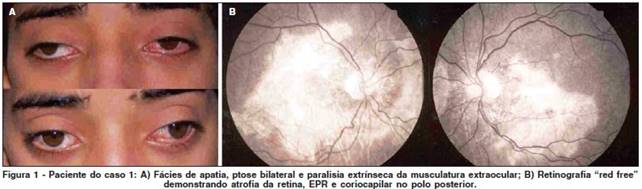
LA SINDROME DI KEARNS SAYRE KSS (Kearns–Sayre Syndrome ) è una grave malattia ad insorgenza sporadica caratterizzata dalla triade: 1) Oftalmoplegia Esterna Progressiva (PEO) con ptosi (abbassamento) palpebrale bilaterale; 2) Retinopatia Pigmentaria; 3) insorgenza prima dei 20 anni. Segni aggiuntivi frequenti sono l’incoordinazione motoria (atassia) di origine cerebellare, il deterioramento mentale, la sordità, e alterazioni del ritmo cardiaco. Vi è spesso ritardo della crescita.
Alterazioni quantitative del mtDNA
Una riduzione della quantità del mtDNA è comunemente chiamata “deplezione”. Questa alterazione si associa comunemente a delle forme ad insorgenza infantile ad andamento progressivo. Gli organi più spesso coinvolti sono: muscolatura scheletrica e cardiaca, fegato e cervello. Le deplezioni del mtDNA sono causate da mutazioni in geni nucleari (vedi capitolo “mitocondriopatie dovute a mutazioni in geni nucleari”).
Mutazioni puntiformi del mtDNA.
Si tratta di quadri clinici associati a sostituzioni di singole basi o a micro-inserzioni/micro-delezioni, nella molecola del mtDNA. Queste mutazioni possono interessare sequenze codificanti RNA transfer (tRNA), RNA ribosomali (rRNA), o RNA messaggeri di proteine mitocondriali (mRNA). A differenza dei riarrangiamenti, che sono per lo più sporadici, quasi tutte le mutazioni puntiformi vengono trasmesse per via matrilineare. Spesso, ma non sempre, queste mutazioni sono eteroplasmiche. Sebbene, ad oggi, centinaia di mutazioni puntiformi siano state descritte in associazione con uno spettro estremamente eterogeneo di presentazioni cliniche, le mutazioni di gran lunga più frequenti sono solo quattro, e sono associate a sindromi cliniche piuttosto ben definite.
La Encefalomiopatia Mitocondriale con Acidosi Lattica ed episodi simil-Stroke (Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes MELAS), (OMIM540000) è definita dalla presenza delle seguenti manifestazioni: 1) episodi di tipo ictale (stroke-like) causati da lesioni cerebrali focali spesso localizzate nelle aree parieto-occipitali; 2) acidosi lattica o comunque livelli anormali di lattato nel sangue (e liquor); 3) fibre “ragged-red” nella biopsia muscolare. Altri segni di coinvolgimento del sistema nervoso centrale comprendono il deterioramento mentale, la cefalea ricorrente con vomito “cerebrale”, epilessia focale o generalizzata, e sordità neurosensoriale. La malattia è trasmessa per via materna e l’esordio è variabile, dalla primissima infanzia all’età giovanile-adulta.
La sindrome MELAS è tipicamente associata alla mutazione A3243G, nel gene codificante il tRNA per la Leucina (codone UUR). Sono state in seguito riportate altre mutazioni puntiformi associate a MELAS, anche se si tratta di casi più rari.
La mioclono epilessia con fibre rosse sfilacciate (Myoclonic Epilepsy and Ragged Red Fibers MERRF),
(OMIM545000) è caratterizzata dall’associazione di mioclono, epilessia, debolezza ed ipotrofia muscolare, incoordinazione motoria (atassia), e, talvolta, deterioramento mentale. L’entità delle manifestazioni cliniche può essere estremamente variabile nell’ambito della stessa famiglia. Tale variabilità si ritiene sia in relazione alla quantità di mtDNA mutato rispetto al normale (eteroplasmia) ed alla variabilità nella distribuzione tissutale della mutazione. La maggior parte delle famiglie affette è portatrice della transizione A8344G, nella sequenza del tRNA per la lisina.
Numerose altre mutazioni puntiformi del mtDNA sono state associate a diversi fenotipi clinici in singoli pazienti o in poche famiglie.
Il DNA mitocondriale è una parte di DNA di piccole dimensioni situata in ogni mitocondrio. Esso codifica per 13 acidi ribonucleici messaggeri (mRNA). due RNA ribosomiali (rRNA) e 22 RNA di trasporto ed è trasmesso. unicamente delle madri, a tutti i loro figli. Normalmente la maggior parte degli individui ha un solo tipo di DNA mitocondriale, ma possono essere presenti diversi tipi di DNA mitocondriale per tessuto il che spiega per esempio la variabilità dei fenotipi tra fratelli, che in teoria dovrebbero essere colpiti nella loro totalità.
Diverse sono le mutazioni descritte: la prima e la più frequente. la mutazione A1555G dell’RNA ribosomiale 12S(1). quindi le mutazioni C1494T dell’RNA ribosomiale l2S e T75llC,T7510C dell’RNA di trasferimento della senna.
L’ipoacusia dovuta alla mutazione A1555G può essere di ogni grado e apparire a qualunque età, spontaneamente o dopo assunzione di aminoglicosidi(2). Infatti il bersaglio degli aminoglicosidi è l’RNA ribosomiale batterico e la forma dell’RNA ribosomiale 12S con mutazione A1555G diviene simile agli rRNA batterici che legano gli aminoglicosidj. Questo spiega la comparsa di ipoacusia per dosi normali di aminoglicosidi in questi pazienti. Anche la mutazione 12SC1494T può indurre una suscettibilità agli arninoglicosidi (3).
1, Kubisch C. Schroeder BC. Friedrich T. Lutjohann B. EL-Amraoui A. Mania S. et al, KCNQ4. a novel potassium channel expressed in sensory outei hair celis. is mutated in dominant deafness Cell 96:437- 46.1999
2. Khanim F. Kirk J. Latif F. Barrett TG WFS 1/Wolfram in mutations. Wolfram syndrome. and associated diseases Hum Mutat 17:357-67. 2001
3. Young TL, lves E, Lynch E. Person R. Snook 5. MacLaren L. et al Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram sindrome gene WFSI. Hum Mol Genet 10.2509-14. 2001
FORME SINDROMICHE
Forme sindromiche. Le forme più frequenti di ipoacusie sindromiche (Tab II) associate a mutazioni del DNA mitocondriale comprendono sindromi neuromuscolari mitocondriali acquisite come la KSS (Kearns–Sayre Syndrome ). la MERRF (Myoclonic Epilepsy and Ragged Red Fibers), la MELAS (Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes), nonché il diabete mellito e l’ipoacusia ad ereditarietà materna detta anche MIDD (Maternally Inherited Diabetes mellitus and Deafness). Accanto a riarrangiamenti di grandi dimensioni che coinvolgono diversi geni, tutte le mutazioni mitocondriali che conducono a forme di ipoacusia sindromiche sono mutazioni puntiformi nei geni tRNA: non sono state ad oggi ancora identificate mutazioni puntiforrni in geni che codificano per le proteine.
La Kearns–Sayre Syndrome (KSS) è caratterizzata da oftalmoplegia esterna progressiva (PEO) e retinopatia prima dei 20 anni. atassia, blocco cardiaco o successivo aumento delle proteine nel liquor. L’ ipoacusia neurosensoriale può svilupparsi come parte del fenotipo.
L’epilessia mioclonica con fibre rosse sfilacciate (MERRF) è caratterizzata da mioclono, epilessia e atassia. ma anche demenza. atrofia ottica e sordità sono frequenti. Il grado di perdita uditiva è variabile.
L’encefalopatia mitocondriale. acidosi lattica ed episodi simil-ictus (MELAS) fanno parte del quadro di una malattia infantile caratterizzata da vomito intermittente. debolezza degli arti prossimali e ricorrenti insulti cerebrali simili ad ictus che causano emiparesi e cecità corticale. MELAS è spesso associata a bassa statura. L’ampia gamma di segni cimici e sintomi includono perdita dell’udito in circa il 30% delle persone colpite.
Diabete ed ipoacusia neurosensoriale, eredidati materialmente (MIDD) sono stati descritti con diabete mellito e di ipoacusia neurosensoriale in alcune famiglie con mutazioni del DNA mitocondriale, La mutazione più frequente è la 3243A->G (anche nella MELAS). Negli studi di popolazione di diabetici. la mutazione 3243A-> G è stata trovata in una piccola percentuale di pazienti (31. 32).
Tab. II FORME sindromiche da mutazioni di geni mitocondriali (mtDNA)*
|
Sindrome |
Modalità di trasmissione |
Gene |
mutazione |
Principali segni |
|
MELAS |
Mitocondriale Mutazioni puntiformi* |
MTND6 MTTQ
|
3243A>G |
Lesione neuromuscolare lpoacusia retrococleare tardiva, acidosi lattica. diabete |
|
Kearns-Sayre |
Mitocondriale Riarrangiamenti del DNA mitocondriale (delezioni)** |
Delezione mitocondriali |
|
Oftalmoplegia, retinite pigmentosa, lesione neuromuscolare. cardiomiopatia iperproteinorrachia
|
|
MIDD Sordità-diabete (Ballinger-Wallace) |
Mitocondriale |
MTTL1 MTTK |
|
Diabete |
|
MERRF |
Mitocondriale Mutazioni puntiformi* |
MTTL1 MTTH
|
8344A->G 8356T->C |
Lesione neuromuscolare, ritardo psicomotorio, acidosi lattica e piruvica
|
|
Cheratodermia palmoplantare—sordità |
Mitocondriale |
MTTS1 |
|
Cheratodermia |
* eredità matrilineare
** forme quasi sempre sporadiche
SINDROME DI KEARNS SAYRE KSS (Kearns–Sayre Syndrome ).
RIASSUNTO
Definizione
Epidemiologia
Segni e sintomi
STORIA NATURALE
EZIOLOGIA
DIAGNOSI
TERAPIA
RIASSUNTO
La sindrome di Kearns-Sayre è una malattia neuromuscolare, multisistemica mortale, molto rara caratterizzata dall’insorgenza, prima dei 20 anni, di oftalmoplegia, ptosi e retinite pigmentosa. Sono stati descritti più di 200 casi. La prevalenza è stimata tra 1 e 3/100.000. La malattia spesso esordisce con i sintomi oculari caratteristici e evolve con la comparsa progressiva di altri segni correlati alla distribuzione tissutale del difetto molecolare. I sintomi più frequenti sono la sordità, il coinvolgimento cardiaco (cardiomiopatia, difetti della conduzione cardiaca blocco di branca sinistra o di conduzione intracardiaca difetto 1), la miopatia dei muscoli scheletrici, i disturbi intestinali, i deficit ormonali (ipoparatiroidismo, diabete mellito 2 ) e l’insufficienza renale. La malattia ha un’evoluzione lenta, con la comparsa di nuovi sintomi e il lento peggioramento dei sintomi già presenti. Alcuni casi della sindrome di Pearson (si veda questo termine) sono evoluti nella sindrome di Kearns-Sayre. La sindrome è dovuta alla delezione di grosse porzioni del DNA mitocondriale. Le delezioni sono eteroplasmiche, cioè le molecole delete possono coesistere nella cellula con le molecole normali. I sintomi sono presenti solo se la percentuale del DNA mutato è significativa. La soglia dipende dall’organo; corrisponde a circa il 60% nei muscoli scheletrici striati. La maggior parte dei casi di sindrome di Kearns-Sayre è sporadica. Infatti, le delezioni del DNA mitocondriale solo eccezionalmente sono ereditate in maniera verticale per via materna. La diagnosi viene suggerita dal quadro clinico e dalla presenza delle caratteristiche alterazioni morfologiche nei muscoli scheletrici (le fibre che presentano una proliferazione mitocondriale o ‘ragged red fibres’ e le fibre deficitarie di citocromo-C-ossidasi). Può essere confermata dall’individuazione di una percentuale significativa di DNA mitocondriale deleto in un tessuto clinicamente o morfologicamente affetto (di solito i muscoli scheletrici). La diagnosi differenziale si pone con le malattie che presentano quadri simili, come la sindrome di Pearson o l’oftalmoplegia cronica. Il trattamento è sintomatico. La prognosi dipende essenzialmente dal numero degli organi interessati. La malattia evolve lentamente nel corso di decenni.
SINDROME DI KEARNS SAYRE KSS (Kearns-Sayre Syndrome).APPROFONDIMENTO
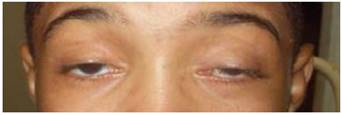
Definizione
E un disturbo multisistemico caratterizzato costantemente dalla triade: esordio entro i 20 anni, oftalmoplegia esterna progressiva e degenerazione pigmentaria della retina, più almeno uno dei seguenti: blocco cardiaco completo, proteine nel CSF >100 mg/dl e atassia cerebellare.
Delezioni su ampia scala del DNA mitocondriale eteroplasmico sono spesso evidenziate nel muscolo scheletrico (raramente in altri tessuti) (Scriver et al., 2001. The Metabolic and Molecular Bases of Inherited Disease, 8th Ed., pp. 2261-74). Caratteristiche supplementari associate con KSS possono includere miopatia, distonia, anomalie endocrine (es. diabete, ritardo di crescita/bassa statura, ipoparatiroidismo), sordità neurosensoriale bilaterale, demenza, cataratta, e acidosi renale tubulare prossimale. Perciò, KSS può colpire molti sistemi organici, KSS e una malattia rara. Esiste una marcata eterogeneità, e sono stati osservati vari tipi di eredita. Anche se KSS probabilmente riduce la spettanza di vita, non esistono dati numerici disponibili. La morbidità dipende dalla gravita e dal numero di sistemi od organi coinvolti che varia grandemente da paziente a paziente. Il blocco cardiaco e una causa significativa e prevenibile di mortalità. KSS non ha predilezione nota di razza o di sesso. Parte della sua caratterizzazione e l’esordio in individui con meno di 20 anni (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
Epidemiologia
Frequenza
Internazionale
Sindrome di Kearns-Sayre è una malattia rara. Sono state osservate eterogeneità Contrassegnato e vari tipi di ereditarietà. Nel 1992, gli autori avevano descritto 226 casi.
Due studi hanno fornito informazioni congruente sulla prevalenza di grandi delezioni mitocondriali nella popolazione adulta. Remes et al hanno stimato una prevalenza di 1,6 casi ogni 100.000 abitanti in una popolazione finlandese (6 pazienti, solo 3 dei quali soddisfaceva i criteri clinici per la sindrome di Kearns-Sayre). [7] Schaefer et al hanno stimato una prevalenza di 1,17 casi per 100.000 abitanti di grandi delezioni mitocondriali Nord Italia; tuttavia, la percentuale di pazienti con sindrome di Kearns-Sayre non è indicato. [8]
Mortalità / morbilità
Anche se probabilmente la sindrome di Kearns-Sayre riduce l’aspettativa di vita, non ci sono dati numerici sono disponibili. La morbidità dipende dalla gravità e dal numero di sistemi o organi coinvolti, che ampiamente varia da paziente a paziente. Blocco cardiaco è una causa importante e prevenibile di mortalità.
Razze
Sindrome di Kearns-Sayre non ha conosciuto predilezione razziale.
Sesso
Sindrome di Kearns-Sayre non ha conosciuto predilezione di sesso.
Età
Parte della caratterizzazione della sindrome di Kearns-Sayre è insorgenza nei soggetti di età inferiore ai 20 anni.
Segni e sintomi
Sintomi: Debolezza muscolare (riduzione cronica e progressiva dei movimenti oculari e ptosi, disfagia, debolezza muscolare scheletrica), disfunzione del CNS (atassia; demenza, encefalopatia, o entrambi; sordita, cecita notturna), Cardiopatia (sincope), Sintomi di disfunzione endocrina.
Segni: Debolezza muscolare (ptosi, oftalmoplegia esterna, diminuita forza muscolare scheletrica), Disfunzione del CNS (retinite pigmentosa, atassia cerebellare, ridotte funzioni mentali superiori, cataratta), Disfunzione cardiaca (bradicardia, insufficienza cardiaca congestizia), Disfunzione endocrina [bassa statura (38% degli individui affetti), ipogonadismo (20% degli individui affetti)] (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
Una disfunzione cardiaca, soprattutto disturbi di conduzione ed aritmie, può verificarsi in qualsiasi momento durante il corso della malattia e può condurre a morte improvvisa. Si può documentare sottoslivellamento del tratto ST all’ECG senza coronaropatia. Bisogna allertare i pazienti su sintomi quali palpitazioni, sensazione di testa vuota, e dispnea e dovrebbero essere esaminati regolarmente da un cardiologo. L’impianto di un pacemaker può risultare salvavita. Può svilupparsi anche un’insufficienza cardiaca causata da una cardiomiopatia congestizia senza difetti di conduzione. Le anomalie neurologiche includono nistagmo, atassia, perdita di udito e demenza. L’esame istologico puo rivelare degenerazione spongiforme della corteccia cerebrale, dei nuclei della base e del tronco cerebrale. Le calcificazioni intracraniche sono comuni. Il contenuto proteico del fluido cerebrospinale e elevato. Disturbi endocrini comprendono ritardo di crescita, ipogonadismo, diabete mellito, tireopatie, ipoparatiroidismo e iperaldosteronismo (Albert M. D., Jakobiec F. A., 2000.)s Editions, pp. 4067-8). ![]()
STORIA NATURALE
Si tratta di una miopatia lentamente progressiva che coinvolge primariamente (e spesso e limitata a) i muscoli oculari estrinseci. Di solito i sollevatori delle palpebre sono i primi ad essere colpiti, dando ptosi, seguita da oftalmoparesi progressiva. Una volta cominciata, la malattia progredisce inesorabilmente finche gli occhi sono immobili. Il coinvolgimento simultaneo di tutti i muscoli extraoculari fa si che gli occhi rimangano in una posizione centrale, cosi che strabismo e diplopia non sono comuni. Quando il paziente tenta di sollevare le sue palpebre e di vedere al di sotto, la testa viene gettata all’indietro ed il muscolo frontale viene contratto, corrugando la fronte (facies hutchinsoniana). Le palpebre sono anormalmente sottili a causa dell’atrofia dei muscoli sollevatori. I muscoli orbicolari dell’occhio sono spesso coinvolti insieme agli extraoculari. Cosi, nell’oftalmoplegia esterna progressiva, come nella miastenia grave e nella distrofia miotonica, c’è una combinazione caratteristica di debolezza nella chiusura e nell’apertura dell’occhio, una combinazione che e quasi sempre miopatica. Gli altri muscoli facciali, masseteri, sternocleidomastoidei, deltoidi, o peronei sono deboli in misura variabile e compromessi circa nel 25% dei casi (Victor M., Ropper A.H., 2001. Adams and Victor’s Principles of Neurology, 7th edition, McGraw-Hill, p. 1502).
KSS e una malattia progressiva, e la prognosi per i pazienti che ne soffrono e infausta. La morte e comune nella terza o quarta decade di vita (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
EZIOLOGIA
KSS compare in seguito a delezioni nel DNA mitocondriale (mtDNA) in grado di indurre un particolare fenotipo. Il gene nel quale avvengono le delezioni e identificato come Online Mendelian Inheritance in Man numero 530000. La comprensione di alcuni aspetti di genetica mitocondriale e importante per capire KSS. L’mtDNA differisce dal DNA nucleare per molti versi. Il genoma mitocondriale di 16.5-kilobasi (kb) e circolare. Il genoma contiene 13 geni strutturali che codificano per peptidi che sono tutti componenti dei complessi della catena respiratoria, e contiene geni che codificano per l’RNA transfer e per l’RNA ribosomiale mitocondriale. Le anomalie ereditari dell’mtDNA dimostrano ereditarietà materna perché durante la formazione dello zigote tutti i mitocondri vengono dall’uovo. Inoltre, ogni cellula contiene centinaia di mitocondri. In certe malattie, inclusa KSS, l’mtDNA mostra eteroplasmia, cioè un misto di mtDNA wild-type e mutante all’interno di una stessa cellula. Il rapporto DNA mutante/selvaggio diviene importante nel determinare il fenotipo in una malattia mitocondriale. L’mtDNA continua a replicare, anche in una cella che non si sta dividendo e ciò può far si che la forma mutata si accumuli nei tessuti non in divisione. Come conseguenza del comune coinvolgimento nelle malattie mitocondriali, anche le velocita relative di replicazione dell’mtDNA mutante e non mutante possono essere un importante fattore patogenetico dei disturbi mitocondriali. Il DNA mutante sembra accumularsi primariamente nei tessuti non in divisione. Non tutti i geni necessari per la funzione mitocondriale si trovano all’interno dell’mtDNA; alcuni sono contenuti all’interno del DNA nucleare. Siccome le malattie mitocondriali colpiscono la funzione della catena respiratoria, ci si può aspettare che abbiano l’effetto massimo su cellule o sistemi organici con le maggiori richieste di energia (es. cervello, muscolo scheletrico e cardiaco, organi di senso, reni). Nei pazienti con KSS si verificano delezioni nell’mtDNA, la maggior parte delle quali sono sporadiche e si crede che accadano come mutazioni della cellula germinale o molto presto nello sviluppo del nuovo embrione. Le delezioni variano per dimensioni (1.3-8 kb) e posizione all’interno del genoma mitocondriale; comunque, il singolo sito più comune e tra le posizioni 8469 e 13147 sul gene (deletion hotspot). Questa mutazione di 4.9-kb incide per un terzo dei casi di KSS. Delezioni si trovano in tutti i tessuti, e di quando in quando al posto delle delezioni si riscontrano duplicazioni in tandem del DNA. Le duplicazioni possono condurre alla malattia attraverso la formazione di delezioni. Anche se la taglia della delezione varia, le delezioni producono un fenotipo simile. Com’e possibile che un gruppo eterogeneo di delezioni mitocondriali possa condurre ad un fenotipo simile? Il meccanismo proposto e basato sulla conoscenza che la trascrizione dell’mtDNA e policistronica, che vuol dire che tutti i geni codificati sui filamenti pesanti e leggeri sono trascritti come 2 grandi filamenti precursori di RNA. Questi poi clivano in filamenti di RNA separati, che comprendono filamenti di RNA transfer. Una delezione in qualsiasi punto del genoma mitocondriale puo alterare la trascrizione o la traduzione di geni che non erano stati interessati dalla delezione. Una identica delezione e stata identificata in pazienti con 2 altre condizioni, cioè la sindrome di Pearson (che comprende anemia sideroblastica dell’infanzia, pancitopenia e insufficienza esocrina pancreatica) e l’oftalmoplegia esterna cronica progressiva (CPEO, che comprende oftalmoplegia esterna, ptosi aponeurogenica bilaterale ed una lieve miopatia prossimale). Le delezioni mitocondriali nella CPEO tendono ad essere localizzate nel tessuto muscolare. Ne le dimensioni ne la collocazione della delezione da sole determinano il fenotipo clinico. Invece, il fenotipo sembra essere determinato dalle quantità relative di mtDNA deleto o selvaggio. E probabile che livelli molto alti di mtDNA deleto in tutti i tessuti provochino la sindrome di Pearson, nella quale la caratteristica dominante e la pancitopenia. Livelli più bassi di mtDNA deleto provocano KSS. Nella CPEO, mtDNA deleti possono essere svelati solamente nel tessuto muscolare. Le eccezioni esistono, e coloro che sopravvivono ad una crisi pancitopenia della sindrome di Pearson possono sviluppare anche KSS. Differenze nel contenuto di DNA mutante si verificano anche in tessuti ed organi diversi. Oltre all’accumulo dell’mtDNA mutato (cioè deleto) nei tessuti postmitotici, si può avere anche la segregazione vegetativa (cioè la segregazione dell’mtDNA della cellula parentale in divisione tra le sue 2 cellule figlie), e questa segregazione può essere disuguale. Tanji e collaboratori suggerirono che la deconnessione delle cellule di Purkinje a livello di nucleo dentato può avere un ruolo nella patogenesi dell’atassia cerebellare in pazienti con KSS; comunque, lo studio investigava solamente 2 pazienti con KSS. Harvey e Barnett ipotizzarono che i cambiamenti spongiformi (visti spesso in tutto il cervello) possono essere responsabili della bassa statura. Test dinamici endocrini indicano che le ghiandole pituitarie di pazienti con KSS sono responsive all’ormone rilasciante le gonadotropine (GnRH); da cui si deduce che il difetto nell’asse ipofisi-gonadi e a livello ipotalamico (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
Diagnosi
La diagnosi e essenzialmente clinica.
Studi di laboratorio: I livelli sierici di creatinin-chinasi possono essere normali o moderatamente elevati. I livelli ematici di lattato e piruvato di solito sono elevati. Nel CSF, il livello di lattato e elevato, anche se i livelli di lattato nel sangue sono nella norma. KSS eleva il livello delle proteine nel CSF. Anche se un test di reazione polimerasica a catena eseguito su DNA da campioni ematici può portare alla scoperta di delezioni nell’mtDNA, il modo migliore per raggiungere la diagnosi definitiva e l’analisi di un campione bioptico muscolare, con quantificazione del livello di delezione utilizzando un’analisi in Southern blot. Si raccomanda uno screening per escludere le anomalie endocrinologiche che si presentano in molti pazienti. I metodi di screening possono includere test per misurare il glucosio sierico, la funzione tiroidea, calcio e magnesio, e i livelli degli elettroliti sierici. Una combinazione di livelli alti di sodio e bassi di potassio può suggerire un iperaldosteronismo, che colpisce il 3% dei pazienti con KSS.
Studi di imaging: L’MRI del cervello ha limitato uso diagnostico. I rilievi MRI possono essere normali o mostrare atrofia cerebrale e cerebellare. I reperti MRI T2-pesati possono mostrare lesioni della sostanza bianca sottocorticale (con o senza coinvolgimento simmetrico) con alto segnale d’intensità nel tronco cerebrale, nel globo pallido, nel talamo e nel cervelletto, da soli o in combinazione. I deficit neurologici e i rilievi MRI correlano poco.
Altri test: L’ECG rivela difetti nella conduzione cardiaca (intervallo PR). L’elettroretinografia aiuta a stimare la degenerazione retinica. L’audiometria aiuta a svelare la sordità neurosensoriale.
Procedure: Eseguendo una puntura lombare si possono misurare i livelli di lattato e le proteine nel CSF. L’esame di una biopsia muscolare puo mostrare fibre rosse raggiate usando una colorazione tricromica di Gomori in singolo passaggio. Le fibre rosse raggiate hanno anormali aggregati di mitocondri subsarcolemmali. I risultati dell’istochimica muscolare rivelano deficienza di citocromo c ossidasi in queste cellule. Comunque, fibre rosse raggiate si osservano anche in biopsie muscolari campionate da altri disturbi mitocondriali e non sono specifiche di KSS. Reperti istologici: In pazienti con KSS, come in pazienti con altre encefalopatie mitocondriali, cambiamenti degenerativi spongiformi si hanno sia nella sostanza grigia che bianca del cervello. La maggior parte delle alterazioni nella sostanza bianca si verificano nel cervello e nel cervelletto; la maggior parte delle alterazioni nella sostanza grigia si verificano nel tronco cerebrale. La perdita neuronale e evidente nel tronco cerebrale e nel cervelletto con demielinizzazione. Depositi di calcio si accumulano nel globo pallido e nel talamo. L’istologia cardiaca mostra anomalie del sistema di conduzione. Grandi mitocondri con struttura anormale si sviluppano sia nel muscolo scheletrico sia nel cardiaco (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
Altri test
- ECG rivela difetti di conduzione cardiaca; è indicato misura dell’intervallo PR. [15]
- L’ecocardiografia è utilizzata per cercare cardiomiopatia.
- Elettroretinografia aiuta a valutare la degenerazione retinica.
- Audiometria consente di rilevare la sordità neurosensoriale.
- Elettroencefalografia durante periodo di encefalopatia rivela attività generalizzata onde lente.
- Reperti elettromiografici e conduzione nervosa possono essere normali o possono mostrare lieve miopatia con o senza neuropatia.
Procedure
- Eseguire una puntura lombare e misurare proteine e lattato livelli nel liquido cerebrospinale.
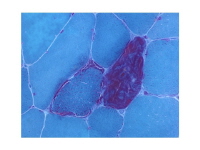
- La biopsia muscolare può rivelare fibre rosse sfilacciate (come mostrato nell’immagine qui sotto).Modificato macchia Gomori Trichrome mostrando fibre rosse sfilacciate. Queste mostrano colorazione rosso tondo la periferia così come all’interno del sarcoplasma, dando un aspetto maculato. Delle due fibre muscolari coinvolte qui raffigurato, quella a destra mostra un più estremo grado di proliferazione mitocondriale e quello di sinistra .anche alcune degenerazione / vacuolizzazione.
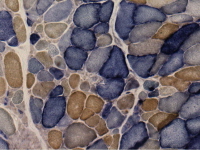
- Istochimica muscolare (come mostrato nell’immagine qui sotto) rivela carenza di citocromo c ossidasi. Il muscolo scheletrico macchiato sia citocromo ossidasi (COX) e succinico deidrogenasi (SDH), due enzimi della catena respiratoria mitocondriale. Fibre che macchiano solo per SDH e sono COX-negative appaiono blu.Original ingrandimento X 50.
I risultati istologici
- Nei pazienti con sindrome di Kearns-Sayre, come con altre encefalopatie mitocondriali, alterazioni degenerative spugnosi avvengono sia la materia grigia e bianca del cervello. Bianco questione spongiosi è prominente negli emisferi cerebrali e tratti di fibre del tronco cerebrale in sindrome di Kearns-Sayre. [16] la perdita di materia grigia è visto anche nel tronco encefalico e strato di cellule di Purkinje. I depositi di calcio si accumulano nel globo pallido e il talamo.
- Studi istologici del cuore mostrano anomalie del sistema di conduzione. Grandi mitocondri con struttura anomala sviluppano in entrambi i muscoli scheletrici e cardiaci.
Diagnosi prenatale e prevenzione
La capacita di effettuare test diagnostici prenatali e predire accuratamente il fenotipo e limitata. I livelli delle mutazioni dell’mtDNA eteroplasmico nei villi coriali o negli amniociti possono non riflettere accuratamente i livelli nei tessuti dei pazienti. Inoltre, la consulenza genetica e difficile a causa della complessità della genetica di KSS e degli altri disturbi correlati (disturbi della funzione mitocondriale). Se viste nell’insieme, la maggior parte delle malattie del sistema di fosforilazione ossidativa sono trasmesse in modo autosomico recessivo. I test sull’mtDNA (Southern blot, analisi di mutazione puntiforme, sequenziamento) possono essere utili nel definire se la causa più probabile per la malattia sia il DNA nucleare o il mitocondriale. Questi approcci possono migliorare di molto l’accuratezza della consulenza genetica.
Ecco le raccomandazioni per una consulenza genetica su malattie causate da mutazioni dell’mtDNA.
I. Eredita materna di una mutazione dell’mtDNA
A. Test sull’albero genealogico: I membri della linea materna dell’albero genealogico sono a rischio di ereditare la mutazione dell’mtDNA. Data la rapida segregazione delle mutazioni dell’mtDNA, la conferma genetica della mutazione dovrebbe essere effettuata in ogni individuo a rischio.
B. Prognosi: Le previsioni basate sul genotipo del paziente su singolo tessuto (% mtDNA mutante vs % mtDNA normale) sono generalmente inattendibili tranne che per individui che albergano alti livelli di mutazioni dell’mtDNA. Uno screening clinico periodico degli organi comunemente coinvolti e necessario per la diagnosi precoce ed il trattamento delle manifestazioni di malattia.
C. Test prenatali: L’esperienza con i test prenatali per le mutazioni dell’mtDNA e limitata. La variabilità del fenotipo dei pazienti con piccole oscillazioni nelle quantità di mtDNA normale rende impossibili accurate previsioni sul fenotipo. La prognosi può essere espressa solamente se il feto e essenzialmente omoplasmico per una mutazione altamente patogenetica dell’mtDNA, che sia eteroplasmico nella madre.
II. Mutazioni spontanee dell’mtDNA
A. Test sull’albero genealogico: A causa dell’alto tasso replicativo delle cellule ematiche, se non si riesce a svelare la mutazione nel sangue non si puo escludere la presenza di mutazioni in altri tessuti. Un tessuto con basso potenziale replicativo come il muscolo scheletrico deve essere esaminato per confermare l’assenza di trasmissione della mutazione dell’mtDNA. Le categorie delle mutazioni spontanee dell’mtDNA meglio caratterizzate sono le delezioni e le duplicazioni. Le delezioni sono in genere confinate alle linee cellulari somatiche, con risparmio della trasmissione alle cellule germinali. E possibile anche che si verifichino mutazioni puntiformi spontanee dell’mtDNA.
B. Prognosi: Vale quanto detto al paragrafo I B. (Scriver et al., 2001. The Metabolic and Molecular Bases of Inherited Disease, 8th Ed., p. 2390).
Diagnosi differenziale
- Atassia identificati da difetti genetici e biochimici
- Blocco atrioventricolare, Second Degree
- Blocco atrioventricolare, Terzo Grado, acquisita
- Oftalmoplegia esterna Cronica Progressive
- Difetto di crescita
- Sindrome MELAS
- Miastenia Gravis
- Sindrome di Pearson
- Retinite Pigmentosa
- Ipoacusia neurosensoriale
TERAPIA
Non esiste alcuna terapia in grado di modificare la storia della malattia in KSS. In futuro, un potenziale trattamento nei pazienti con KSS può tentare di inibire la replicazione dell’mtDNA mutante o stimolare la replicazione dell’mtDNA selvaggio. I problemi associati con KSS vanno trattati convenientemente (es. insulina per il diabete mellito).
Tutti i pazienti con KSS richiedono la cura di un oftalmologo. Può rendersi necessaria una consulenza con un cardiologo riguardo all’impianto di un pacemaker per blocco di conduzione. Consulenze supplementari (es. endocrinologo, neurologo) possono essere necessarie, a seconda dello stato del paziente e della presenza di complicazioni. L’esercizio fisico può aiutare i pazienti con miopatia. Esercizi che inducano accorciamento concentrico dei muscoli portano alla proliferazione di cellule satelliti, i precursori delle cellule muscolari che sono anche coinvolti nella rigenerazione del muscolo. Le cellule satelliti contengono livelli indosabili di mtDNA mutante; se proliferano, la proporzione mtDNA selvaggio/mutante può aumentare con conseguente beneficio. Compiere esercizi fino a raggiungere questo traguardo e difficile per i pazienti gravemente colpiti o giovani. La somministrazione di coenzima Q10 (CoQ10) e di supplementi vitaminici si e dimostrata efficace in casi individuali, anche se gli effetti sono transitori (Posner E., 2002. Kearns-Sayre Syndrome. eMedicine).
Il CoQ10 e un chinone liposolubile che contiene una catena laterale di 10 unita isoprenoidi. Il CoQ10 funziona in modo da trasferire elettroni dal complesso I al complesso III e dal complesso II al complesso III. Questo composto può stabilizzare anche i complessi di fosforilazione ossidativa all’interno della membrana interna e può servire come un potente antiossidante per i radicali liberi dell’ossigeno, anche alle concentrazioni fisiologiche. Nell’uomo, le concentrazioni più alte di coenzima Q si trovano in cuore, fegato, rene e pancreas. Composti usati per il trattamento di supporto di KSS per i quali ci mancano dati consistenti sono: due composti della vitamina K, menadione e fillochinone, somministrati in abbinata con la vitamina C per donare elettroni direttamente al citocromo c; succinato, un composto intermedio del ciclo degli acidi tricarbossilici che dona direttamente elettroni al complesso II; tiamina, nicotinamide e riboflavina (Scriver et al., 2001. The Metabolic and Molecular Bases of Inherited Disease, 8th Ed., pp. 2391-2).
Sindrome di MERRF Myoclonic Epilepsy with Ragged-Red Fibers (MERRF) — Sindrome Fukuhara–epilessia mioclonica con fibre rosse sfilacciate—mioencefalopatia/ malattia delle fibre rosse sfilacciate
Che cosa è la Sindrome di MERRF?
Quanto è diffusa la MERRF?
Quali sono i cambiamenti genetici relativi alla MERRF?
come si fa la diagnosi della sindrome MERRF?
Come fanno le persone ad ereditare la MERRF?
MERRF(APPROFONDIMENTO)
Che cosa è la Sindrome di MERRF?
Per Sindrome di MERRF in campo medico si intende un quadro clinico complesso da difetto mitocondriale.
L’Epilessia mioclonica con fibre rosse sfilacciate (MERRF) è un disturbo che colpisce molte parti del corpo, in particolare i muscoli e il sistema nervoso. Nella maggior parte dei casi, i segni ed i sintomi di questa malattia compaiono durante l’infanzia o l’adolescenza. Le caratteristiche della MERRF variano ampiamente tra gli individui affetti, anche tra i membri della stessa famiglia.
MERRF è caratterizzato da contrazioni muscolari (mioclono), debolezza (miopatia), e la rigidità progressiva (spasticità). Quando le cellule muscolari delle persone colpite sono macchiati e visualizzati al microscopio, queste cellule di solito appaiono anomale. Queste cellule muscolari anomali sono chiamate fibre rosse sfilacciate. Altre caratteristiche di MERRF includono crisi ricorrenti (epilessia), difficoltà di coordinazione dei movimenti (atassia), una perdita di sensibilità alle estremità (neuropatia periferica), e lento deterioramento della funzione intellettuale (demenza). Le persone con questa condizione possono anche sviluppare la perdita dell’udito o ottica atrofia, che è la degenerazione (atrofia) di cellule nervose che trasportano informazioni visive dagli occhi al cervello. Gli individui affetti hanno talvolta bassa statura e una forma di malattia cardiaca nota come cardiomiopatia. Meno comunemente, le persone con MERRF sviluppano tumori grassi, chiamati lipomi, appena sotto la superficie della pelle.
Quanto è diffusa la MERRF?
MERRF è una condizione rara; la sua prevalenza non è nota. MERRF è parte di un gruppo di condizioni note come i disordini mitocondriali, che colpiscono circa 1 a 5.000 persone in tutto il mondo.
Quali sono i cambiamenti genetici relativi alla MERRF?
Le mutazioni nel gene MT-TK sono la causa più comune di MERRF, che si verificano in più di 80 per cento di tutti i casi. Meno frequentemente, le mutazioni nei geni MT-TL1, MT-TH, e MT-TS1 sono stati segnalati per causare i segni e sintomi di MERRF. Le persone con mutazioni nel MT-TL1, MT-TH, o gene MT-TS1 genere hanno segni e sintomi di altri disturbi mitocondriali così come quelli di MERRF.
La MT-TK, MT-TL1, MT-TH, e MT-TS1 geni sono contenuti nel DNA mitocondriale (mtDNA). I mitocondri sono strutture all’interno delle cellule che utilizzano ossigeno per convertire l’energia dal cibo in una forma cellule possono utilizzare attraverso un processo chiamato fosforilazione ossidativa.Sebbene la maggior parte del DNA è confezionato in cromosomi all’interno del nucleo, mitocondri hanno anche una piccola quantità di DNA proprio. I geni associati alla MERRF forniscono istruzioni per fare molecole chiamate trasferimento RNA, che sono cugini chimiche del DNA. Queste molecole aiutano assemblare blocchi di proteine chiamate aminoacidi in full-length, proteine funzionanti all’interno dei mitocondri. Queste proteine eseguono le fasi di fosforilazione ossidativa.
Mutazioni che causano MERRF compromettere la capacità dei mitocondri per produrre proteine, utilizzare l’ossigeno, e produrre energia. Queste mutazioni particolarmente colpiscono organi e tessuti con requisiti di alta energia, come il cervello e muscoli. I ricercatori non hanno determinato come i cambiamenti nel mtDNA portano a segni e sintomi di MERRF specifici.
Una piccola percentuale di casi MERRF sono causate da mutazioni in altri geni mitocondriali, e in alcuni casi la causa della condizione è sconosciuta.
Per saperne di più sulla MT-TH , MT-TK , MT-TL1 , e MT-TS1 geni e del DNA mitocondriale .
Consulta l’elenco dei geni associati alla MERRF.
Come si fa la diagnosi della sindrome MERRF?
La diagnosi della sindrome MERRF basa sulla dimostrazione di accumulo anomalo di lattato nel sangue o, più spesso, nel liquido cerebrospinale, e sulla biopsia muscolare, che rivela la presenza di fibre muscolari negative citocromo c ossidasi e fibre rosse sfilacciate. Le analisi biochimiche del muscolo mostra spesso citocromo c ossidasi carenza o combinato difetti della catena respiratoria. Eteroplasmia (cioè la convivenza della forma mutante con una popolazione residua di tipo selvatico DNA mitocondriale) dovrebbe essere presa in considerazione durante l’identificazione della causale. La proporzione della mutazione può variare notevolmente tra i tessuti. Tuttavia, nella sindrome MERRF, questa percentuale è spesso molto elevata (superiore al 90%) in tutti i tessuti e la mutazione può quindi essere studiata nel sangue. L’eteroplasmia rende la consulenza genetica molto arduo nella sindrome MERRF
Come fanno le persone ad ereditare la MERRF?
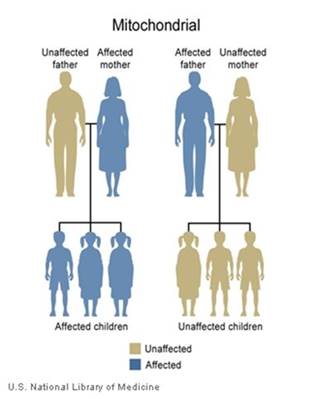 MERRF è ereditata in un modello mitocondriale, che è anche conosciuto come eredità materna.Questo modello di ereditarietà si applica a geni contenuti nel mtDNA. Poiché le cellule uovo, ma non le cellule spermatiche, contribuiscono mitocondri nello sviluppo embrionale, solo le femmine passano condizioni mitocondriali ai loro figli. Malattie mitocondriali possono apparire in ogni generazione di una famiglia e può colpire sia i maschi che le femmine, ma i padri non passare tratti mitocondriali ai loro figli.
MERRF è ereditata in un modello mitocondriale, che è anche conosciuto come eredità materna.Questo modello di ereditarietà si applica a geni contenuti nel mtDNA. Poiché le cellule uovo, ma non le cellule spermatiche, contribuiscono mitocondri nello sviluppo embrionale, solo le femmine passano condizioni mitocondriali ai loro figli. Malattie mitocondriali possono apparire in ogni generazione di una famiglia e può colpire sia i maschi che le femmine, ma i padri non passare tratti mitocondriali ai loro figli.
Nella maggior parte dei casi, le persone con MERRF ereditano un gene mitocondriale alterato dalla madre, che può o non può mostrare sintomi della malattia. Meno frequentemente, i risultati di disturbo una nuova mutazione in un gene mitocondriale e si verifica nelle persone con una storia familiare di MERRF.
Dove posso trovare le informazioni circa la diagnosi o la gestione di MERRF?
Queste risorse riguardano la diagnosi o la gestione di MERRF e possono includere fornitori di trattamento.
- Recensione Gene: MERRF

- Test genetici Registry: Mioclono con epilessia con fibre rosse sfilacciate
- Kennedy Krieger Institute: Disturbi mitocondriali
- MedlinePlus Enciclopedia: Lipoma
- MedlinePlus Enciclopedia: nervo Atrofia ottica
- Encyclopedia MedlinePlus: neuropatia periferica
- MitoAction: Suggerimenti e strumenti per vivere con Mito
- United malattia mitocondriale Fondazione: trattamenti e terapie
Si potrebbe anche trovare informazioni sulla diagnosi o la gestione di MERRF in risorse educative e di supporto del paziente .
Informazioni generali sul diagnosi e la gestione delle malattie genetiche è disponibile nel Manuale.Per saperne di più test genetici , in particolare la differenza tra test clinici e test di ricerca .
Dove posso trovare informazioni generali sulle malattie genetiche?
Il manuale fornisce le informazioni di base sulla genetica in un linguaggio chiaro.
- Che cos’è il DNA mitocondriale?
- Icambiamenti nel DNA mitocondriale possono influenzare la salute e lo sviluppo?
- Quali sono i diversi modi in cui una condizione genetica può essere ereditata?
- Se una malattia genetica corre nella mia famiglia, quali sono le probabilità che i miei figli avranno la condizione?
MERRF(APPROFONDIMENTO)
Sinonimo: epilessia mioclonica Associata con fibre Ragged Red
Salvatore DiMauro, MD e Michio Hirano, MD.
Pubblicazione iniziale: 3 giugno 2003; Ultimo aggiornamento: 18 agosto, 2009.
Riassunto
Caratteristiche della malattia. MERRF (epilessia mioclonica con fibre rosse sfilacciate) è una malattia multisistemica caratterizzata da mioclono, che è spesso il primo sintomo, seguita da epilessia generalizzata, atassia, debolezza, e la demenza. L’esordio è di solito durante l’infanzia, che si verifica dopo il normale sviluppo iniziale. Risultati più comuni sono la perdita dell’udito, bassa statura, atrofia ottica e cardiomiopatia con Wolff-Parkinson-White (WPW). Retinopatia pigmentosa e lipomatosi Occasionalmente si osservano.
Diagnosi / testing La diagnosi clinica di MERRF si basa sulle quattro caratteristiche “canonici” seguenti: Mioclono, epilessia generalizzata, atassia, e fibre rosse sfilacciate (RRF) nella biopsia muscolare. Il mitocondriale DNA (mtDNA) gene codifica MT-TK tRNA Lys è il gene più comunemente associati con MERRF. Il più comune mutazione , presente in oltre l’80% dei colpiti individui con reperti tipici, è una transizione A-G al nucleotide 8344 (m.8344A> G). Le mutazioni sono di solito presenti in tutti i tessuti e sono convenientemente rilevati nel mtDNA da leucociti di sangue. Tuttavia il verificarsi di ” dell’eteroplasmia “in disturbi del mtDNA può determinare una variazione distribuzione tissutale di mtDNA mutato. Quindi, in individui con pochi sintomi compatibili con MERRF o in asintomatici parenti materni di un individuo affetto, la mutazione patogena può essere rilevabile nel mtDNA da leucociti e può essere rilevata solo in altri tessuti, come i fibroblasti cutanei, sedimento urinario, mucosa orale , follicoli piliferi, o, più affidabile, muscolo scheletrico.
Gestione Trattamento delle manifestazioni: convenzionali farmaci antiepilettici (AED) per le convulsioni;terapia fisica per migliorare qualsiasi funzione motoria; esercizio aerobico; terapia farmacologica standard per sintomi cardiaci. Levetiracetam, clonazepam, zonisamide, e acido valproico (VPA) sono stati utilizzati per il trattamento dell’epilessia mioclonica; tuttavia, VPA può provocare carenza di carnitina secondaria e deve essere evitato o utilizzata con supplementazione di L-carnitina.
Altro: coenzima Q 10 (100 mg 3x/giorno) e L-carnitina (1000 mg 3x/giorno) sono spesso utilizzati nella speranza di migliorare la funzione mitocondriale.
La consulenza genetica. MERRF è causata da mutazioni nel mtDNA e si trasmette per eredità materna. Il padre di un probando non è a rischio per avere il mtDNA-malattia che causa la mutazione . La madre di un probando ha di solito la mutazione mitocondriale e può o non può avere sintomi. Un maschio con una mutazione mtDNA non può trasmettere la mutazione su qualsiasi sua progenie. Una femmina con la mutazione (sia influenzata o inalterato ) trasmette la mutazione di tutta la sua prole. La diagnosi prenatale per MERRF è possibile se una mutazione del mtDNA è stato rilevato nella madre. Tuttavia, poiché il carico mutazionale nei tessuti della madre e nei tessuti fetali nel campione (cioè, amniociti e villi coriali) potrebbe non corrispondere a quello di altri tessuti fetali e beuse il carico mutazionale nei tessuti campione prenatale potrebbe spostarsi in utero o dopo la nascita secondaria di casuale mitotico segregazione , la previsione del fenotipo da studi prenatali non è possibile.
Diagnosi
Diagnosi clinica
La diagnosi clinica di MERRF (m yoclonic e pilepsy con r agged r Ed F IBERS) si basa su quattro caratteristiche “canoniche” seguenti:
- Miocloni
- Epilessia generalizzata
- Atassia
- Fibre rosse sfilacciate (RRF) nella biopsia muscolare
Ulteriori manifestazioni frequenti sono i seguenti:
- Ipoacusia neurosensoriale
- Miopatia
- Neuropatia periferica
- Demenza
- Bassa statura
- Intolleranza all’esercizio
- Atrofia ottica
Segni clinici meno comuni (osservati in <50% dei colpiti individui) sono i seguenti:
- Cardiomiopatia
- Retinopatia pigmentosa
- Segni piramidali
- Oftalmoparesi
- Lipomi multipli
Test
L’acidosi lattica sia nel sangue e nel liquor. Nei soggetti con MERRF, le concentrazioni di lattato e piruvato sono comunemente elevati a riposo e aumentare eccessivamente dopo l’attività moderata.
Nota: Altre situazioni (non correlati alla diagnosi di MERRF o di altre malattie mitocondriali), in cui lattato e piruvato può essere elevato sono eventi neurologici acuti quali il sequestro o ictus.
Concentrazione proteica CSF elevata. La concentrazione di proteina CSF può essere aumentata, ma raramente supera 100 mg / dL.
Elettroencefalogramma (EEG) mostra di solito generalizzate spike e onda scarichi con sfondo rallentando, ma gli scarichi epilettiformi focali può anche essere visto.
Elettrocardiogramma mostra spesso pre-eccitazione; blocco cardiaco non è stato descritto.
Elettromiografia (EMG) e la velocità di conduzione nervosa (NCV) studi sono coerenti con una miopatia, ma neuropatia possono coesistere.
MRI del cervello mostra spesso atrofia del cervello e gangli basali calcificazione. Bilaterale necrosi putaminal e atrofia del tronco cerebrale e del cervelletto sono stati riportati [ Orcesi et al 2006 , Ito et al 2008].
La biopsia muscolare mostra tipicamente fibre rosse sfilacciate (RRF) con la colorazione tricromica di Gomori modificato e fibre iperattivi con la succinato deidrogenasi (SDH) macchia. Sia RRF e alcuni non-RRF non riescono a macchiare con la reazione istochimica per la citocromo c ossidasi (COX). Di tanto in tanto, RRF non può essere osservato [ Mancuso et al 2007 ].
Gli studi catena respiratoria. Analisi biochimica degli enzimi della catena respiratoria in estratti muscolari di solito spettacoli ridotta attività dei complessi della catena respiratoria contenenti subunità codificate dal mtDNA, carenza particolarmente COX. Tuttavia, studi biochimici possono anche essere normale.
Test Genetico Molecolare
Gene
- Il mitocondriale DNA (mtDNA) gene MT-TK codifica tRNA lisina (tRNA Lys) è il gene più comunemente associati con MERRF.
- Un individuo con la MERRF fenotipo aveva la mutazionem.611G> A in MT-TF, che codifica la fenilalanina tRNA (tRNA Phe) [ Mancuso et al 2004 ].
- Un paziente ha presentato con MERRF e retinopatia pigmentosa causata dal m.15967G> Unamutazione in MT-TP [ Blakely et al 2009 ].
Sperimentazione clinica
Analisi di mutazione mirata Quattro mutazioni MT-TK. (m.8344A> G, m.8356T> C, m.8363G> A e m.8361G> A, vedi Tabella 1 e Tabella 3 ) rappresentano circa il 90% delle mutazioni in individui con MERRF.
- Il più comune mutazione in MERRF, presente in oltre l’80% dei colpiti individui con reperti tipici, è m.8344A> G.
- Tre ulteriori mutazioni, m.8356T> C, m.8363G> A, e m.8361G> A, sono presenti nel 10% deicolpiti individui.
Nota: Le mutazioni sono di solito presenti in tutti i tessuti e possono essere rilevati in mtDNA da leucociti del sangue in soggetti con tipica MERRF; Tuttavia il verificarsi di ” dell’eteroplasmia “in disturbi del mtDNA può determinare una variazione distribuzione tissutale di mtDNA mutato. Quindi, in individui con pochi sintomi compatibili con MERRF o nei parenti materni asintomatici, il patogeno mutazione può essere rilevabile in mtDNA dai leucociti e può essere rilevata solo in altri tessuti, come ad esempio colture di fibroblasti cutanei, sedimento urinario, mucosa orale (da collutorio) , follicoli piliferi, o, più affidabile, muscolo scheletrico.
Scansione mutazione / analisi di sequenza . Il restante 10% dei colpiti individui probabilmente hanno altre mutazioni nel mtDNA, compreso il m.611G> Una mutazione in MT-TF e la m.15967G> Una mutazione inMT-TP (vedi Diagnosi differenziale ).
Nota: la scansione Mutation / analisi di sequenza è utilizzato per rilevare mutazioni tutta mtDNA e non è specifico per MERRF.
Tabella 1. Summary of Molecular Genetic Testing Usato in MERRF
|
Gene Symbol |
Metodo di prova |
Mutazioni identificate |
L’identificazione della mutazione frequenza con il metodo di prova 1 |
|
MT-TK |
m.8344A> G |
> 80% |
|
|
m.8356T> C |
~ 10% |
||
|
m.8363G> A |
|||
|
m.8361G> A |
|||
|
MT-TF |
Scansione mutazione /analisi di sequenza |
m.611G> A |
<5% 2 |
|
MT-TP |
m.15967G> A |
||
|
mtDNA |
Scansione mutazione /analisi di sequenza |
Varianti Sequenza 3 |
90% -95% 4 |
4. Scansione Mutation / analisi di sequenza è utilizzato per rilevare mutazioni tutta mtDNA e non è specifico per MERRF. Il generale mutazione tasso di rilevamento per MERRF mediante analisi di scansione / sequenza di mtDNA è del 90% -95%.
1. La capacità del metodo di prova utilizzato per rilevare una mutazione che è presente nel indicata gene
2. La proporzione di MERRF causata da queste due mutazioni
. 3 Esempi di mutazioni identificate da analisi di sequenza possono includere piccole delezioni intrageniche / inserimenti e missense, nonsense, e sito di splice mutazioni; di solito, parziale, intere-o multigeniche delezioni / duplicazioni non vengono rilevati.
Interpretazione dei risultati delle prove
- Per le questioni da considerare nell’interpretazione di analisi di sequenza dei risultati, fare clic qui .
- Gli individui pensato di avere MERRF ma senza un identificabile MT-TK mutazione possono ospitare il MT-TF mutazione m.611G> A , la MT-TP mutazione m.15967G> A transizione, o un’altra mutazione del mtDNA (vedi Diagnosi differenziale ).
Strategia di sperimentazione
Stabilire la diagnosi di un probando
- Tipicamente, leucociti nel sangue il DNA è inizialmente proiettato per la m.8344A> Gmutazioneseguita dalla proiezione del m.8356T> C , m.8363G> A , e m.8361G> A mutazioni. In alternativa, il DNA dalla mucosa buccale, muscolo, o sedimento urinario possono essere sottoposti a screening per le mutazioni del mtDNA.
- Se si escludono le mutazioni MT-TK, mtDNA sequenziamento può essere eseguita in probandi con storie familiari compatibili con ereditarietà materna. In casi simplex (cioè, una singola occorrenza in una famiglia) con mioclono, epilessia e atassia, biopsia muscolare è spesso utile nel rilevare segni di disfunzione mitocondriale come fibre rosse sfilacciate, fibre ossidasi-deficient citocromo c, o difetti biochimici di mitocondriale enzimi della catena respiratoria.
La diagnosi prenatale per gravidanze a rischio richiede la preventiva identificazione della mutazione responsabile della malattia in famiglia.
Geneticamente correlati (alleliche) Disturbi
MT-TK
- m.8344A> G può anche essere associata a isolati miopatia, simile distrofia muscolare dei cingoli , o con più lipomi, di solito si trova nel collo e la zona delle spalle (sindrome Ekbom). Altre presentazioni cliniche della m.8344A> G mutazione includono la degenerazione spinocerebellare esindrome di Leigh o isolato sindrome di Leigh.
- m.8356T> C . In due famiglie con la m.8356T> C mutazione , alcuni colpiti individui avuto tipica MERRF ma altri anche avuto episodi di simil-ictus ed emicrania (MERRF / MELAS si sovrappongono).
- m.8363G> A è stato associato con la tipica MERRF, ma anche con cardiomiopatia o la sindrome di Leigh [ Shtilbans et al 2000 ].
Descrizione clinica
Storia Naturale
MERRF è una malattia multisistemica caratterizzata da mioclono, che è spesso il primo sintomo, seguita da epilessia generalizzata, atassia, debolezza, e la demenza. L’esordio è di solito durante l’infanzia, dopo un normale sviluppo iniziale. Tabella 2 elenca i sintomi ei segni osservati in 62 affetti individui [ Hirano & DiMauro 1996 ]. Circa l’80% (34/42) aveva una storia familiare compatibile con ereditarietà materna, ma non tutti i parenti materni sono stati colpiti e non tutti coloro che sono colpiti avuto il quadro completo MERRF.Ad esempio, sette parenti oligosintomatici avevano “dei cingoli miopatia” come l’unica manifestazione. La depressione può essere una caratteristica sotto-riconosciuta di MERRF [ Molnar et al 2009 ].
Di tanto in tanto gli individui che soddisfano i criteri clinici per MERRF hanno anche colpi (MERRF / MELASsovrappongono) [ Crimi et al 2003 , Melone et al 2004 , Naini et al 2005 ] o oftalmoplegia esterna progressiva e retinopatia, che ricorda la sindrome di Kearns-Sayre [ Nishigaki et al 2003 ].
Maternamente ereditato degenerazione spinocerebellare e la sindrome di Leigh, atipica malattia di Charcot-Marie-Tooth (vedi Charcot-Marie-Tooth panoramica ), e la sindrome di Leigh è stato segnalato come manifestazioni insolite nelle famiglie con individui con diagnosi di MERRF.
Manifestazioni insoliti in individui con la m.8344A> Gmutazione includono:
- Sudden Infant Death Syndrome (SIDS) in una ragazza bambino che aveva una cardiomiopatia insospettato con le caratteristiche istologiche di cardiomiopatia istiocitoide [ Vallance et al 2004 ]
- Disfonia spasmodica in una donna di 46 anni con altrimenti abbastanza tipico personale e la storia familiare di MERRF [ Peng et al 2003 ]
- Riposo dolore muscolare come il sintomo iniziale nei bambini [ van de Glind et al 2007 ]
- Mioclono, epilessia e atassia senza fibre rosse sfilacciate [ Mancuso et al 2007 ]
- Parkinsonismo, neuropatia e miopatia [ Horvath et al 2007 ]
- Infantile-insorgenza atassia, mioclono, e bilaterale necrosi putaminal sul cervello MRI [ Orcesi et al 2006 ]
- Insufficienza respiratoria improvvisa in età adulta [ Wiedemann et al 2008 ]
- Malattie demielinizzanti del sistema nervoso centrale e periferico acuta [ Erol et al 2009 ].
A sei-anno-vecchio ragazzo con il m.8631G> Amutazione sequestri sviluppati e mioclono, seguita da atassia, deficit cognitivo e la perdita dell’udito neurosensoriale. Parenti materni erano oligosintomatico [ Rossmanith et al 2003 ].
Un individuo con la MT-TF m.611G> Unamutazione aveva tronco mite e prossimale debolezza degli arti, atassia cerebellare, bilaterale segno di Babinski, e frequenti scosse miocloniche [ Mancuso et al 2004 ].
Tabella 2. Segni e sintomi, osservati in 62 individui con MERRF
|
Segno / Sintomo |
Presente / Evaluated |
Percentuale |
|
Miocloni |
62/62 |
100% |
|
Epilessia |
62/62 |
100% |
|
Normale sviluppo iniziale |
17/17 |
100% |
|
RRF (fibre rosse sfilacciate) |
47/51 |
92% |
|
La perdita dell’udito |
41/45 |
91% |
|
L’acidosi lattica |
24/29 |
83% |
|
Cronaca di famiglia |
34/42 |
81% |
|
Intolleranza all’esercizio |
8/10 |
80% |
|
Demenza |
39/52 |
75% |
|
Neuropatia |
17/27 |
63% |
|
Bassa statura |
4/7 |
57% |
|
Sensazione deteriorate |
9/18 |
50% |
|
Atrofia ottica |
14/36 |
39% |
|
Cardiomiopatia |
2/6 |
33% |
|
Sindrome di Wolff-Parkinson-White |
2/9 |
22% |
|
Retinopatia pigmentosa |
4/26 |
15% |
|
Segni piramidali |
Gli 8/60 |
13% |
|
Oftalmoparesi |
3/28 |
11% |
|
Lipomatosi |
2/60 |
3% |
Genotipo-fenotipo Correlazioni
Nessuna chiara correlazione è stata individuata tra genotipo e clinica fenotipo per colpiti gli individui, né è chiaro il motivo tipico MERRF è associata a mutazioni in MT-TK.
Per tutte le mutazioni del mtDNA, espressione clinica dipende da tre fattori:
- Eteroplasmia. L’abbondanza relativa di mtDNA mutante
- Distribuzione tissutale di mtDNA mutante
- Effetto soglia. La vulnerabilità di ogni tessuto per il metabolismo ossidativo alterato
La soglia di vulnerabilità dei tessuti, probabilmente non varia sostanzialmente tra gli individui, ma carico mutazionale variabile e distribuzione tissutale può spiegare la diversità clinica delle persone con MERRF.
La vulnerabilità selettiva del nucleo dentato del cervelletto e il nucleo olivary del midollo è inspiegabile. Anche inspiegabile è la patogenesi delle molteplici lipomi tipicamente associati a mutazioni in MT-TK.
Penetranza
Vedere genotipo-fenotipo Correlazioni .
Anticipazione
Nessuna prova di anticipazione è stata trovata, ma la conoscenza del difetto molecolare può favorire la diagnosi precoce nelle generazioni successive.
Nomenclatura
Ramsay Hunt [1921] ha descritto sei individui con un disturbo caratterizzato da atassia, mioclono ed epilessia, che chiamò “dissinergia cerebellaris myoclonica.” Gli individui con diagnosi di sindrome di Ramsay Hunt dovrebbero essere indagati per MERRF.
Prevalenza
Tre studi epidemiologici di malattie mtDNA-correlati in Nord Europa hanno concordemente basse stime per la prevalenza del m.8344A> G mutazione :
- 0-1.5:100,000 nella popolazione adulta del nord della Finlandia [ Remes et al 2005 ]
- 0.25:100,000 nella popolazione adulta del nord dell’Inghilterra [ Chinnery et al 2000 ]
- 0-0.25:100,000 in una popolazione pediatrica di Svezia occidentale [ Darin et al 2001 ]
Vedere malattie mitocondriali Panoramica per informazioni generali prevalenza.
Diagnosi differenziale
. Segni neurologici La diagnosi differenziale include:
- Altri disturbi mitocondriali (vedi mitocondriale Disturbi Panoramica )
- Disturbi POLG legati . Un individuo con mioclono, epilessia, atassia, neuropatia periferica, ma non ci sono fibre rosse sfilacciate aveva autosomiche recessive mutazioni in POLG, che codifica per la subunità catalitica della mitocondriale DNA polimerasi gamma [ Van Goethem et al 2003 ].
- MERRF / MELAS sovrapposte sindrome causata da mutazioni in MT-ND5, che possono inizialmente assomigliare MERRF ( Tabella 3 ) [ Crimi et al 2003 , DiMauro & Davidzon 2005 ,Naini et al 2005 ].
- A MERRF / sindrome di Kearns-Sayre (KSS), la sindrome di sovrapposizione causata da una MT-TL1 mutazione ; riportato in un individuo [ Nishigaki et al 2003 ]
- Sindromi caratterizzate da atassia (ad esempio, DRPLA ) (vedi anche atassia ereditaria Panoramica) e epilessia mioclonica, tra cui la malattia Unverricht-Lundborg , malattia di Lafora , neuronale Ceroidolipofuscinosi , e sialidosi [ Zupanc e Legros 2004 ].
Il coinvolgimento multisistemico, acidosi lattica, la prova di eredità materna, e la biopsia muscolare con RRF (fibre rosse sfilacciate) MERRF distinguono da altre condizioni.
Lipomi. Altre sindromi che causano lipomi multipli (ad esempio, aerei lipomatosi simmetrica) devono essere considerati.
Gestione
Valutazioni dopo la diagnosi iniziale
Per stabilire l’estensione della malattia in un individuo con diagnosi di MERRF (m yoclonic e pilepsy associato a r agged r Ed F IBERS), si raccomandano le seguenti valutazioni:
- Misura di altezza e peso per valutare la crescita
- La valutazione audiologica
- La valutazione oftalmologica
- Valutazione delle abilità cognitive
- Valutazione Fisioterapia
- Valutazione neurologica, compreso MRI, MRS, e EEG se si sospetta crisi epilettiche
- Valutazione cardiaca
Trattamento delle manifestazioni
Il disturbo sequestro può essere trattata con la terapia anticonvulsivante convenzionale. Studi No controllati hanno confrontato l’efficacia di diversi farmaci anticonvulsivanti.
I mioclono migliorato sostanzialmente in tre dei quattro soggetti trattati con levetiracetam [ Crest et al 2004 ,Mancuso et al 2007 ].
La terapia fisica è utile per tutte le abilità motorie deteriorati.
L’esercizio aerobico è utile in MERRF e altre malattie mitocondriali [ Taivassalo & Haller 2004 ].
La terapia farmacologica standard è usato per trattare i sintomi cardiaci.
Sorveglianza
Rivalutazione ogni sei a 12 mesi per progressione di malattia che possono giustificare una terapia sintomatica (ad esempio, la modifica della terapia anticonvulsivante) è appropriato.
Valutazione dei parenti a rischio
Vedere la consulenza genetica per le questioni relative alle prove di a rischio parenti per consulenza geneticascopi.
Terapie sotto inchiesta
Cerca ClinicalTrials.gov per l’accesso alle informazioni sugli studi clinici per una vasta gamma di malattie e condizioni. Nota: Non ci può essere studi clinici per questo disturbo.
La consulenza genetica
La consulenza genetica è il processo di fornire individui e famiglie, con informazioni sulla natura, eredità, e le implicazioni di malattie genetiche per aiutarli a prendere decisioni personali e mediche informate. La sezione seguente si occupa di valutazione del rischio genetico e l’uso della storia della famiglia e il test genetico per chiarire lo status genetico per i familiari. Questa sezione non è destinata ad affrontare tutte le questioni personali, culturali o etici che gli individui possono affrontare o per sostituire la consultazione con una genetica professionali. -ED.
Modalità di ereditarietà
MERRF è causata da mutazioni nel mtDNA e si trasmette per eredità materna.
Rischio per i membri della famiglia
I genitori di un probando
- Il padre di un probando non è a rischio di avere la mtDNA-malattia che causa la mutazione .
- La madre di un probando (di solito) ha il mtDNA mutazione e può o non può avere sintomi.
- In alternativa, il probando può avere un mitocondriale de novo (somatica) mutazione .
Fratelli e sorelle di un probando
- Il rischio per i fratelli dipende dallo stato genetico della madre.
- Se la madre ha il mtDNA mutazione , tutti i fratelli e sorelle di un probando erediteranno il mtDNA mutazione responsabile della malattia e possono o non possono avere sintomi.
Figli di un probando
- Tutti prole di femmine con un mtDNA mutazione erediterà la mutazione.
- Figli di maschi con un mtDNA mutazione non sono a rischio di ereditare la mutazione.
Altri membri della famiglia di un probando
- Il rischio per gli altri membri della famiglia dipende dallo stato genetico dei probando madre s ‘.
- Se la madre ha un mtDNA mutazione , i suoi fratelli e la madre sono a rischio.
Genetica correlati Counseling Problemi
Variabilità fenotipica. Il fenotipo di un individuo con una mtDNA mutazione risultato di una combinazione di fattori quali la gravità della mutazione, la percentuale di mitocondri mutanti (carico mutazionale), e gli organi e tessuti in cui si trovano (distribuzione tissutale). Diversi membri della famiglia spesso ereditare diverse percentuali di mtDNA mutante e quindi possono avere una vasta gamma di sintomi clinici.
Interpretazione dei risultati dei test di membri asintomatici a rischio familiari è estremamente difficile.Pronostico fenotipo sulla base dei risultati del test non è possibile. Inoltre, l’assenza del mtDNA mutazione in un tessuto (ad esempio, sangue) non garantisce che la mutazione è assente in altri tessuti.
Pianificazione familiare
- Il momento ottimale per la determinazione del rischio genetico e discussione della disponibilità di test prenatale è prima della gravidanza. Allo stesso modo, le decisioni circa il test per determinare lo stato genetico dei membri a rischio familiari asintomatici sono fatti meglio prima della gravidanza.
- È opportuno offrire consulenza genetica (compresa la discussione generale dei rischi potenziali per la prole e le opzioni riproduttive) ai giovani adulti che sono affetti oa rischio; tuttavia, non è possibile prevedere specifici sul potenziale gravità della malattia nella prole.
Bancario DNA è la conservazione di DNA (tipicamente estratto da cellule bianche del sangue) per un possibile uso futuro. Poiché è probabile che la metodologia di analisi e la nostra comprensione dei geni, mutazioni e malattie miglioreranno in futuro, si dovrebbe considerare il DNA bancario di affetti individui.
Test prenatale
Sebbene i risultati di diagnosi prenatale per MERRF non possono fornire ulteriori informazioni, è possibile mediante analisi del DNA estratto da cellule fetali ottenute da amniocentesi (generalmente realizzate in ~ 15-18 settimane di gestazione) o prelievo dei villi coriali (generalmente realizzate in ~ 10-12 settimane di gestazione). Il mtDNA specifica mutazione nella madre deve essere identificato prima della diagnosi prenatale può essere eseguita.
Nota: l’età gestazionale è espresso in settimane mestruali calcolati sia dal primo giorno dell’ultimo periodo mestruale normale o da misure ad ultrasuoni.
Interpretazione dei risultati diagnostici prenatali è complessa per i seguenti motivi:
- Il carico mutazionale nei tessuti della madre e nei tessuti fetali campione (cioè, amniociti e villi coriali) potrebbe non corrispondere a quello di altri tessuti fetali.
- Pronostico fenotipo , età di insorgenza, la gravità, o la velocità di progressione non è possibile.
Risorse
Personale GeneReviews ha selezionato le seguenti organizzazioni e / o sostegno umbrella specifici di malattia e / o registri per il beneficio di individui con questo disturbo e le loro famiglie. GeneReviews non è responsabile per le informazioni fornite da altre organizzazioni. Per informazioni sui criteri di selezione, clicca qui .
- United mitocondriale Disease Foundation (UMDF)
8085 Salisburgo Strada
Suite 201
Pittsburg PA 15239
Telefono: 888-317-8633 (numero verde); 412-793-8077
Fax: 412-793-6477
Email: info@umdf.org
- Distrofia Muscolare – USA (MDA)
3300 East Sunrise Unità
Tucson AZ 85718
Telefono: 800-572-1717
Email: mda@mdausa.org
- Registro Nazionale Malattie oftalmica genotipizzazione Network – eyeGENE ®
Telefono: 301-435-3032
Email: eyeGENEinfo@nei.nih.gov
- RDCRN Patient Registry Contatto: North American Consorzio malattia mitocondriale
Genetica molecolare
Informazioni nelle tabelle Genetica Molecolare e OMIM può differire da quella nel resto del GeneReview: tavoli possono contenere informazioni più recenti. – ED.
Tabella A. MERRF: Geni e database
|
Gene Symbol |
Cromosomica Locus |
Proteine Nome |
|
MT-TK |
Non applicabile |
|
|
MT-TF |
Non applicabile |
|
|
MT-TP |
Non applicabile |
I dati sono compilati dai seguenti riferimenti standard: Simbolo gene da HGNC ; cromosomico luogo, nome luogo, regione critica, complementazione gruppo da OMIM ; Nome proteine UniProt . Per una descrizione di basi di dati (Locus Specific, HGMD) per cui sono previsti collegamenti, fare clic qui .
Tabella B. OMIM Inserzioni per MERRF ( Visualizza tutto in OMIM )
|
Epilessia mioclonica ASSOCIATA fibre rosse sfilacciate; MERRF |
|
|
TRASFERIMENTO RNA mitocondriale, LYSINE; MTTK |
|
|
TRASFERIMENTO RNA mitocondriale, FENILALANINA; MTTF |
|
|
TRASFERIMENTO RNA mitocondriale, PROLINE; MTTP |
Patogenesi Genetica Molecolare
L’origine delle mutazioni del mtDNA è incerto. E ‘anche chiaro come le mutazioni puntiformi del mtDNA causano MERRF. Utilizzando rho 0 linee cellulari (linee permanenti cellulari umane svuotate del loro mtDNA dall’esposizione al bromuro di etidio) ripopolati con i mitocondri che ospitano il m.8344A> G mutazione ,Chomyn et al [1991] ha trovato che i carichi mutazionali elevati correlati con diminuzione della sintesi proteica, è diminuito consumo di ossigeno, e citocromo c ossidasi carenza. I polipeptidi contenenti numeri elevati di residui di lisina sono stati più gravemente colpiti dalla mutazione, suggerendo che la mutazione MT-TK inibisce direttamente la sintesi proteica. Analogamente, miotubi in coltura contenenti più dell’85% mtDNA mutante mostrato diminuito traduzione , in particolare di proteine contenenti un gran numero di residui di lisina. Cellule che ospitano la mutazione m.8344A> G contenuta diminuzione dei livelli di tRNA Lys e aminoacylated tRNALys. Inoltre, la mutazione m.8344A> G bloccato una modifica del tRNA Lys, con conseguente ridotta sintesi proteica [ Yasukawa et al 2001 ]. La mutazione sembra essere funzionalmente recessiva perché solo circa il 15% di tipo selvatico mtDNA ripristina traduzione e citocromo c ossidasi attività a livelli quasi normali.
Masucci et al [1995] hanno confermato che la sintesi proteica e il consumo di ossigeno sono diminuiti in rho 0cellule ripopolata con mtDNA ospitare sia il m.8344A> G o m.8356T> C mutazione , e ha individuato aberrante della proteina mitocondriale in entrambe le linee cellulari, che essi attribuiti a ribosomiale frame-shifting. Studi di ingegnerizzati in vitro trascritti mutanti tRNA Lys ha dimostrato che le mutazioni associate a MERRF avuto alcun effetto sull’efficienza lysylation considerando che le due mutazioni associate con encefalomiopatie senza caratteristiche tipiche MERRF (m.8313G> A e m.8328G> A in MT-TK) lysylation gravemente compromessa [ Sissler et al 2004 ].
Varianti alleliche normali. Polimorfismi benigni sono particolarmente frequenti in mtDNA e sono elencati inwww.mitomap.org . MT-TK è l’unica mtDNA gene che codifica tRNA Lys.
Varianti alleliche patologiche. Vedi Tabella 3 .
Tabella 3. Patologica allelica varianti in mitocondriale DNA associati con MERRF
|
Mitocondriale DNA |
Gene Symbol |
Proteina Amino |
Reference Sequence |
|
m.8344A> G |
MT-TK |
Nessuna proteina tradotta |
|
|
m.8356T> C |
|||
|
m.8363G> A |
|||
|
m.8361G> A |
|||
|
m.611G> A |
MT-TF |
||
|
m.15967G> A |
MT-TP |
Vedere Riferimento rapido per una spiegazione di nomenclatura. Le varianti denominate secondo le linee guida attuale nomenclatura ( www. Hgvs.org / )
1. Designazione Variant che non è conforme alle convenzioni di denominazione corrente
Per ulteriori informazioni, vedere Tabella A .
Normale prodotto genico . Il prodotto gene normale della MT-TK e MT-TF (tRNA Lys e tRNA Phe) sono indispensabili per l’integrazione di questi aminoacidi nelle proteine mitocondriali nascenti.
Abnormal prodotto genico . Consulta Molecular Genetic Patogenesi .
Capitolo Notes
Cronologia delle revisioni
- 18 August 2009 (me) Comprehensive update posted live
- 27 September 2005 (me) Comprehensive update posted to live Web site
- 3 June 2003 (ca) Review posted to live Web site
- 8 May 2003 (sdm) Original submission
Copyright © 1993-2014, University of Washington, Seattle. Tutti i diritti riservati.
Per ulteriori informazioni, consultare il Copyright GeneReviews e Disclaimer d’uso .
For questions regarding permissions: ude.wu@tssamda .
Bookshelf ID: NBK1520 PMID: 20301693
MELAS(Mitochondrial Encephalomyopathy, Lactic,Acidosis,and Stroke-like episodes)
Riassunto
Che cosa è la MELAS
Incidenza
Segni e Sintomi
Genetica
Prognosi
MELAS APPROFONDIMENTO medscape
Riassunto
La sindrome MELAS (encefalomiopatia mitocondriale con acidosi lattica e episodi simili a ictus) è una patologia progressiva caratterizzata da disturbi neurologici acuti paragonabili a ischemie cerebrali, associati a iperlactatemia e miopatia mitocondriale. La prevalenza è sconosciuta. Durante l’infanzia o la prima adolescenza, i pazienti sono soggetti di solito a crisi acute, forse causate da un’infezione o da uno sforzo fisico. Queste crisi si associano a cefalee, vomito e a volte ad episodi pseudoischemici, emiparesi e emianopsia. Si manifestano spesso in pazienti con sintomi cronici come deficit motorio, sordità, diabete, bassa statura, cardiomiopatia, ritardo dello sviluppo, difficoltà di apprendimento, di memoria e di attenzione. La malattia è dovuta alle mutazioni del DNA mitocondriale. Sono state identificate 10 mutazioni differenti ma l’80% dei casi è dovuto alla mutazione 3243A>G nel gene del tRNA della leucina (tRNA Leu). Questa mutazione viene definita spesso mutazione MELAS, sebbene sia associata a segni clinici diversi; la sua prevalenza in Europa è stimata in 1 su 6250 e si associa alla sindrome in oltre il 7,5% dei pazienti. La diagnosi si basa sulle manifestazioni cliniche e sulla risonanza magnetica cerebrale. La risonanza magnetica può rivelare la presenza di numerose lesioni profonde nella materia grigia e bianca del cervello, mentre la TAC identifica atrofia cerebrale e calcificazioni dei gangli basali. La risonanza e la TAC mostrano che le lesioni non sono confinate nelle aree vascolari e di conseguenza gli episodi acuti non si possono considerare tipici ictus. La concentrazione anomala di lattato è frequente nel sangue e pressoché costante nel liquido cerebrospinale. La biopsia muscolare è anomala nell’85% dei pazienti, mostra una proliferazione mitocondriale atipica (fibre rosse lacerate) e fibre muscolari con deficit di citocromo-ossidasi. L’analisi delle attività enzimatiche della catena respiratoria muscolare può rivelare un deficit del complesso I o un deficit combinato dei complessi I e IV. L’identificazione della mutazione causale deve tenere conto della sua costante eteroplasmia, cioè la sua coesistenza con una popolazione residuale di DNA mitocondriale normale. La proporzione delle mutazioni varia notevolmente a seconda dei tessuti, ma spesso può essere molto elevata (oltre il 90% della popolazione del DNA mitocondriale) e può essere ricercata anche nel sangue. La terapia è complicata dall’eteroplasmia. La mutazione è trasmessa per eredità materna. Un maschio affetto non è in grado di trasmettere la malattia. Sebbene una proporzione elevata della mutazione nel sangue della madre aumenti il rischio di nascita di un bambino gravemente affetto dalla malattia, esistono molti esempi di segregazione estrema madre-figlio, che rendono difficile la consulenza genetica. La presenza di percentuali eterogenee della mutazione nei diversi tessuti impedisce, in teoria, la diagnosi prenatale. Sono state effettuate poche sperimentazioni cliniche. Una sperimentazione recente ha scoperto che il dicloroacetato ha un effetto deleterio nel medio termine. L’evoluzione spontanea, fatta di crisi seguite da recuperi e ricadute, rende difficile valutare il miglioramento in alcuni pazienti sottoposti a trattamenti (che utilizzano il coenzima Q10 e l’analogo idebenone, la creatina monoidrato e l’arginina) o il peggioramento dovuto ad alcuni trattamenti come l’acido valproico (farmaco a effetto antiepilettico responsabile di episodi simili a ictus). La prognosi è grave. Gli episodi potrebbero provocare il decesso del paziente e la loro ricorrenza nel lungo periodo può causare un deterioramento mentale, perdita della vista e dell’udito e grave miopatia, che potenzialmente può contribuire alla perdita dell’autonomia
Che cosa è la MELAS
L’encefalopatia mitocondriale, acidosi lattica con episodi tipo ictus – abbreviato MELAS
(miopatia, encefalopatia, acidosi lattica, e episodi di simil-ictus) – è una della famiglia di citopatie mitocondriali , che comprendono anche MERRF , e neuropatia ottica ereditaria di Leber , è una malattia neurodegenerativa progressiva caratterizzata da episodi neurologici acuti simili a tratti associati iperlattatemia e miopatia mitocondriale.
È stata per la prima volta caratterizzata con questo nome nel 1984 da Pavlakis SG ET AL.[1] Una caratteristica di queste malattie è che sono causate da difetti del DNA nel genoma mitocondriale che viene ereditata esclusivamente dal genitore femminile[Hirano M, Pavlakis SG,1994].[2]. [ 2 ] Tuttavia, è importante sapere che alcuni delle proteine essenziali per la normale funzione mitocondriale sono prodotte dal genoma nucleare, e vengono successivamente trasportati ai mitocondri per uso.Come tale, le mutazioni in queste proteine possono provocare malattie mitocondriali, ma può essere ereditato da entrambi i genitori maschi e femmine nel modo tipico. La malattia può manifestarsi in entrambi i sessi.
INCIDENZA
La prevalenza esatta della malattia è sconosciuta. I pazienti di solito presentano durante l’infanzia o la prima età adulta con crisi acute, che possono essere attivati da infezione o l’esercizio fisico. Queste crisi socio cefalea, vomito e talvolta segni pseudo-ictus, come confusione, emiparesi e emianopsia. Spesso si verificano in pazienti con sintomi cronici come la debolezza muscolare, sordità, diabete, bassa statura, cardiomiopatia, ritardo dello sviluppo, difficoltà di apprendimento, perdita di memoria o disturbi dell’attenzione.
SEGNI E SINTOMI
MELAS è una condizione che colpisce molti dei sistemi del corpo, in particolare il cervello e il sistema nervoso (encefalopatia) e muscoli (miopatia). Nella maggior parte dei casi, i segni ed i sintomi di questa malattia compaiono durante l’infanzia, dopo un periodo di normale sviluppo. [3] I primi sintomi possono includere debolezza muscolare e dolore, mal di testa ricorrenti, perdita di appetito, vomito e convulsioni. Gli individui più colpiti sperimentano episodi di simil-ictus che iniziano prima dei 40 anni Questi episodi spesso comportano debolezza temporanea muscolare su un lato del corpo (emiparesi), alterazione della coscienza, alterazioni della visione, convulsioni, e forti mal di testa che assomigliano emicranie. Episodi di simil-ictus ripetuti possono progressivamente danneggiare il cervello, con conseguente perdita della vista, problemi di movimento, e una perdita della funzione intellettuale (demenza). Gli episodi di simil-ictus può essere mis-diagnosi di epilessia da un medico non è a conoscenza della condizione MELAS.
La maggior parte delle persone affette da MELAS hanno un accumulo di acido lattico nei loro corpi, una condizione chiamataacidosi lattica . Aumento acidità nel sangue può portare a vomito, dolore addominale, stanchezza estrema (stanchezza), debolezza muscolare, perdita di controllo dell’intestino, e difficoltà di respirazione. Meno comunemente, le persone con MELAS possono sperimentare spasmi involontari muscolari (mioclono), muscolare alterata coordinazione ( atassia ), perdita di udito, problemi cardiaci e renali, diabete, epilessia, e gli squilibri ormonali.
La presentazione di alcuni casi è simile a quello della sindrome di Kearns-Sayre . [4]
La diagnosi di sindrome di MELAS si basa sulla presentazione e il cervello di imaging clinico. La risonanza magnetica può rivelare numerose lesioni iperintense in T2 nella materia bianca e grigia cerebrale, mentre la tomografia computerizzata mostra atrofia cerebrale e calcificazioni dei gangli della base. Essi mostrano che le lesioni non sono confinate ai territori vascolari e quindi che gli episodi acuti non sono tratti tipici. Accumulo anomalo di lattato è frequente nel sangue e quasi costante nel liquido cerebrospinale. La biopsia muscolare è anormale in circa l’85% dei pazienti. Essa mostra proliferazione anormale mitocondriale (fibre rosse sfilacciate) e fibre muscolari con un difetto di citocromo c ossidasi. Analisi del muscolo attività della catena respiratoria può rivelare carenza del complesso o un deficit combinato dei complessi I e IV. Identificazione della mutazione causale deve tenere conto della costante eteroplasmia cioè la sua coesistenza con una popolazione residuale di tipo selvaggio DNA mitocondriale. Proporzioni mutazione può variano notevolmente tra i tessuti, ma è il più delle volte molto elevata (superiore al 90%) e possono quindi essere studiati nel sangue. La consulenza genetica è molto arduo nella sindrome MELAS causa della eteroplasmia. Mutazioni del DNA mitocondriale sono trasmessi secondo ereditarietà materna. Un uomo affetto non può trasmettere la malattia. La mutazione verrà trasmessa lungo la linea materna, ma la sua quota è essenzialmente imprevedibile. Anche se le proporzioni più elevate di mutazione nel sangue del risultato madre in un rischio maggiore di avere un bambino con grave fenotipo, ci sono molti esempi di estrema segregazione della mutazione da madre a figlio, che impediscono la consulenza genetica efficiente a livello individuale. L’eterogeneità possibilità nella proporzione della mutazione tra i tessuti ostacola teoricamente diagnosi prenatale. Pochissimi studi clinici adeguati sono stati condotti con MELAS pazienti. Un recente ha trovato dicloroacetato ad avere effetti negativi nel medio termine. Evoluzione spontanea della malattia con crisi acute, la remissione e ricorrenza rende difficile valutare il miglioramento clinico riportato in alcuni pazienti trattati con MELAS trattamenti di supporto (tra cui il coenzima Q10 e il suo analogo idebenone, la creatina monoidrato e arginina) o l’impatto deleterio di trattamento, come acido valproico (un farmaco antiepilettico riferito di provocare episodi di simil-ictus). La prognosi è scarsa. I pazienti possono morire durante un episodio simil-ictus e, insieme a episodi ricorrenti, spesso sviluppano deterioramento mentale, perdita della vista e dell’udito, nonché grave miopatia, che potrebbe condurre alla perdita di autonomia.
Revisore esperto (s)Dr Anne LOMBES Ultimo aggiornamento: Luglio 2006
Genetica

La biopsia muscolare di una persona con diagnosi di MELAS ma portare nessuna mutazione conosciuta.(A) Modificato colorazione tricromica Gomori mostra diverse fibre rosse sfilacciate (punta di freccia). (B) II fibre, fibre scure e qualche fibre con collezioni anomali di mitocondri (freccia) macchia citocromo c ossidasi mostra Type-1 leggermente macchiato e Type. Nota fibre negative citocromo c ossidasi, come di solito visto in encefalopatia mitocondriale, acidosi lattica e episodi di simil-ictus (MELAS). (C) succinato deidrogenasi colorazione mostra alcuni blu fibre laceri e intensa colorazione nei mitocondri dei vasi sanguigni (freccia). Microscopia (d) Electron mostrando raccolta anomala di mitocondri con inclusioni paracristalline (punta di freccia), inclusioni osmiophilic (grande punta di freccia) e vacuoli mitocondriali (piccola punta di freccia). Abu-Amero et al. Journal of Medical Case Reports 2009. [5]
MELAS è causata da mutazioni nei geni nel DNA mitocondriale.
NADH deidrogenasi
Alcuni dei geni ( MT-ND1 , MT-ND5 ) colpiti in MELAS codificano proteine che sono parte della NADH deidrogenasi (chiamato anche complesso I) in mitocondri, che aiuta a convertire l’ossigeno e zuccheri semplici di energia.
RNA di trasferimento
Altri geni ( MT-TH , MT-TL1 , e MT-TV ) codificano mitocondriali RNA specifiche di trasferimento ( tRNA ).
Mutazioni in MT-TL1 causano più del 80 per cento di tutti i casi di MELAS. Essi compromettono la capacità dei mitocondri per produrre le proteine, usa l’ossigeno, e producono energia. I ricercatori non hanno determinato come i cambiamenti nel portare DNA mitocondriale ai segni e sintomi di MELAS specifici. Essi continuano a studiare gli effetti di mutazioni geniche mitocondriali in diversi tessuti, in particolare nel cervello.
La malattia è causata da mutazioni del DNA mitocondriale. Almeno 10 diverse mutazioni sono state identificate ma 80% dei casi sono dovuti alla 3243A> G mutazione nel gene transfer leucina RNA ( tRNA Leu ). Questa mutazione è quindi spesso definito come la mutazione MELAS nonostante la sua associazione con diverse presentazioni cliniche: la sua prevalenza nella popolazione generale europea è stata stimata in 1/6250. La mutazione 3271T> C nel gene tRNA Leu è associata con la sindrome in un ulteriore 7,5% dei pazienti
Inheritance
Questa condizione è ereditata in un modello mitocondriale, che è anche conosciuto come eredità materna e eteroplasmia . Questo modello di ereditarietà applica ai geni contenuti nel DNA mitocondriale. Poiché le cellule uovo, ma non le cellule spermatiche, contribuiscono mitocondri nello sviluppo embrionale, solo le femmine passano condizioni mitocondriali ai loro figli.Malattie mitocondriali possono apparire in ogni generazione di una famiglia e può colpire sia i maschi che le femmine, ma i padri non passare tratti mitocondriali ai loro figli. Nella maggior parte dei casi, le persone con MELAS ereditano un gene mitocondriale alterato dalla madre. Meno frequentemente, i risultati di disturbo una nuova mutazione in un gene mitocondriale e si verifica nelle persone con una storia familiare di MELAS.
Prognosi
Non vi è alcun trattamento conosciuto per la malattia di base, che è progressiva e fatale. I pazienti sono gestiti secondo quali aree del corpo sono interessate in un momento particolare. Gli enzimi , aminoacidi , antiossidanti e vitamine sono stati utilizzati, ma non ci sono stati successi consistenti segnalati.
Anche se non ci sono stati studi controllati sui benefici a lungo termine di manipolazioni dietetiche, i seguenti supplementi hanno mostrato risultati promettenti e dato speranza ai pazienti MELAS.
· CoQ10 è stato utile per alcuni pazienti MELAS. [6]Nicotinamide è stata utilizzata perché complesso l accetta elettroni da NADH e infine trasferisce elettroni CoQ10.
· Riboflavina è stato segnalato per migliorare la funzione di un paziente con deficit di l complesso e la mutazione 3250T-C. [7]
· La somministrazione di L-arginina nei periodi acuti e interictali può rappresentare una nuova potenziale terapia per questa sindrome per ridurre i danni cerebrali a causa di compromissione della vasodilatazione nelle arterie cerebrali a causa di ossido nitrico esaurimento. [8][9]
· C’è anche un caso in cui è stato utilizzato con successo succinato per trattamento di convulsioni non controllate in MELAS pazienti, anche se questa modalità di trattamento è ancora da indagare a fondo e ampiamente raccomandato.
·
La diagnosi di sindrome di MELAS si basa sulla presentazione e il cervello di imaging clinico. La risonanza magnetica può rivelare numerose lesioni iperintense in T2 nella materia bianca e grigia cerebrale, mentre la tomografia computerizzata mostra atrofia cerebrale e calcificazioni dei gangli della base. Essi mostrano che le lesioni non sono confinate ai territori vascolari e quindi che gli episodi acuti non sono tratti tipici. Accumulo anomalo di lattato è frequente nel sangue e quasi costante nel liquido cerebrospinale. La biopsia muscolare è anormale in circa l’85% dei pazienti. Essa mostra proliferazione anormale mitocondriale (fibre rosse sfilacciate) e fibre muscolari con un difetto di citocromo c ossidasi. Analisi del muscolo attività della catena respiratoria può rivelare carenza del complesso o un deficit combinato dei complessi I e IV. Identificazione della mutazione causale deve tenere conto della costante eteroplasmia cioè la sua coesistenza con una popolazione residuale di tipo selvaggio DNA mitocondriale. Proporzioni mutazione può variano notevolmente tra i tessuti, ma è il più delle volte molto elevata (superiore al 90%) e possono quindi essere studiati nel sangue. La consulenza genetica è molto arduo nella sindrome MELAS causa della eteroplasmia. Mutazioni del DNA mitocondriale sono trasmessi secondo ereditarietà materna. Un uomo affetto non può trasmettere la malattia. La mutazione verrà trasmessa lungo la linea materna, ma la sua quota è essenzialmente imprevedibile. Anche se le proporzioni più elevate di mutazione nel sangue del risultato madre in un rischio maggiore di avere un bambino con grave fenotipo, ci sono molti esempi di estrema segregazione della mutazione da madre a figlio, che impediscono la consulenza genetica efficiente a livello individuale. L’eterogeneità possibilità nella proporzione della mutazione tra i tessuti ostacola teoricamente diagnosi prenatale. Pochissimi studi clinici adeguati sono stati condotti con MELAS pazienti. Un recente ha trovato dicloroacetato ad avere effetti negativi nel medio termine. Evoluzione spontanea della malattia con crisi acute, la remissione e ricorrenza rende difficile valutare il miglioramento clinico riportato in alcuni pazienti trattati con MELAS trattamenti di supporto (tra cui il coenzima Q10 e il suo analogo idebenone, la creatina monoidrato e arginina) o l’impatto deleterio di trattamento, come acido valproico (un farmaco antiepilettico riferito di provocare episodi di simil-ictus). La prognosi è scarsa. I pazienti possono morire durante un episodio simil-ictus e, insieme a episodi ricorrenti, spesso sviluppano deterioramento mentale, perdita della vista e dell’udito, nonché grave miopatia, che potrebbe condurre alla perdita di autonomia”Revisore esperto (s)Dr Anne LOMBES Ultimo aggiornamento: Luglio 2006
MELAS APPROFONDIMENTO medscape
La sindrome MELAS (encefalomiopatia mitocondriale con acidosi lattica e episodi simili a ictus) è una patologia progressiva caratterizzata da disturbi neurologici acuti paragonabili a ischemie cerebrali, associati a iperlactatemia e miopatia mitocondriale. La prevalenza è sconosciuta. Durante l’infanzia o la prima adolescenza, i pazienti sono soggetti di solito a crisi acute, forse causate da un’infezione o da uno sforzo fisico. Queste crisi si associano a cefalee, vomito e a volte ad episodi pseudoischemici, emiparesi e emianopsia. Si manifestano spesso in pazienti con sintomi cronici come deficit motorio, sordità, diabete, bassa statura, cardiomiopatia, ritardo dello sviluppo, difficoltà di apprendimento, di memoria e di attenzione. La malattia è dovuta alle mutazioni del DNA mitocondriale. Sono state identificate 10 mutazioni differenti ma l’80% dei casi è dovuto alla mutazione 3243A>G nel gene del tRNA della leucina (tRNA Leu). Questa mutazione viene definita spesso mutazione MELAS, sebbene sia associata a segni clinici diversi; la sua prevalenza in Europa è stimata in 1 su 6250 e si associa alla sindrome in oltre il 7,5% dei pazienti. La diagnosi si basa sulle manifestazioni cliniche e sulla risonanza magnetica cerebrale. La risonanza magnetica può rivelare la presenza di numerose lesioni profonde nella materia grigia e bianca del cervello, mentre la TAC identifica atrofia cerebrale e calcificazioni dei gangli basali. La risonanza e la TAC mostrano che le lesioni non sono confinate nelle aree vascolari e di conseguenza gli episodi acuti non si possono considerare tipici ictus. La concentrazione anomala di lattato è frequente nel sangue e pressoché costante nel liquido cerebrospinale. La biopsia muscolare è anomala nell’85% dei pazienti, mostra una proliferazione mitocondriale atipica (fibre rosse lacerate) e fibre muscolari con deficit di citocromo-ossidasi. L’analisi delle attività enzimatiche della catena respiratoria muscolare può rivelare un deficit del complesso I o un deficit combinato dei complessi I e IV. L’identificazione della mutazione causale deve tenere conto della sua costante eteroplasmia, cioè la sua coesistenza con una popolazione residuale di DNA mitocondriale normale. La proporzione delle mutazioni varia notevolmente a seconda dei tessuti, ma spesso può essere molto elevata (oltre il 90% della popolazione del DNA mitocondriale) e può essere ricercata anche nel sangue. La terapia è complicata dall’eteroplasmia. La mutazione è trasmessa per eredità materna. Un maschio affetto non è in grado di trasmettere la malattia. Sebbene una proporzione elevata della mutazione nel sangue della madre aumenti il rischio di nascita di un bambino gravemente affetto dalla malattia, esistono molti esempi di segregazione estrema madre-figlio, che rendono difficile la consulenza genetica. La presenza di percentuali eterogenee della mutazione nei diversi tessuti impedisce, in teoria, la diagnosi prenatale. Sono state effettuate poche sperimentazioni cliniche. Una sperimentazione recente ha scoperto che il dicloroacetato ha un effetto deleterio nel medio termine. L’evoluzione spontanea, fatta di crisi seguite da recuperi e ricadute, rende difficile valutare il miglioramento in alcuni pazienti sottoposti a trattamenti (che utilizzano il coenzima Q10 e l’analogo idebenone, la creatina monoidrato e l’arginina) o il peggioramento dovuto ad alcuni trattamenti come l’acido valproico (farmaco a effetto antiepilettico responsabile di episodi simili a ictus). La prognosi è grave. Gli episodi potrebbero provocare il decesso del paziente e la loro ricorrenza nel lungo periodo può causare un deterioramento mentale, perdita della vista e dell’udito e grave miopatia, che potenzialmente può contribuire alla perdita dell’autonomia.
Sfondo
Miopatia mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) La sindrome è una malattia neurodegenerativa progressiva. I pazienti possono presentare sporadicamente o come membri di pedigree materni con un’ampia varietà di presentazioni cliniche. La presentazione tipica dei pazienti con sindrome MELAS include caratteristiche che compongono il nome del disturbo, come encefalomiopatia mitocondriale, acidosi lattica , ed episodi di. Altre caratteristiche, come le convulsioni, diabete mellito , perdita dell’udito, malattie cardiache, bassa statura , endocrinopatie, intolleranza all’esercizio, e disfunzioni neuropsichiatrici sono chiaramente parte del disordine.
Fisiopatologia
Episodi di ictus e miopatia mitocondriale caratterizzano la sindrome MELAS. Coinvolgimento organo multisistemica è visto, compreso il SNC, muscolo scheletrico, occhio, muscolo cardiaco, e, più raramente, la GI e renale. [1]
Circa l’80% dei pazienti con le caratteristiche cliniche della sindrome MELAS hanno un eteroplasmica mutazione puntiforme A-to-G nel ciclo dihydrouridine del RNA di trasferimento (tRNA) Leu (UUR) gene alla coppia di basi (bp) 3243 (vale a dire, 3243 A → G mutazione). [2] Tuttavia, altri DNA mitocondriale (mtDNA) si osservano, tra cui il m.3244 G → A, m.3258 T → C, m.3271 T → C, e m.3291 T → C nel mitocondriale tRNA Leu (UUR) gene.
La patogenesi degli episodi di simil-ictus nella sindrome MELAS non è stato completamente chiarito. [3] Questi episodi ictus metaboliche può essere non vascolari e grazie alla transitoria fosforilazione ossidativa (OXPHOS) disfunzioni all’interno del parenchima cerebrale. Un angiopatia mitocondriale di piccoli vasi è responsabile per migliorare il contrasto delle regioni colpite e alterazioni mitocondriali di cellule endoteliali e cellule muscolari lisce dei vasi sanguigni. La disfunzione multisistemica in pazienti con sindrome MELAS può essere dovuta sia parenchimale e difetti OXPHOS vascolari. Aumento della produzione di radicali liberi in associazione con un difetto OXPHOS che porta a vasocostrizione può compensare l’effetto di potenti vasodilatatori (ad esempio, ossido nitrico).
Gli episodi di simil-ictus inusuali e maggiore morbilità osservata nella sindrome di MELAS può essere secondaria ad alterazioni dell’omeostasi ossido di azoto che causano danni microvascolari. L’ossido nitrico può legare i citocromo c ossidasi-positive siti nei vasi presenti nel SNC sangue, spostando ossigeno eme-legato e conseguente diminuita disponibilità di ossigeno nel tessuto circostante e diminuito ossido nitrico libero. Inoltre, l’accoppiamento della disfunzione mitocondriale vascolare con depressione corticale diffusione potrebbe essere alla base della distribuzione selettiva di lesioni ischemiche nella corteccia posteriore in questi soggetti.
Mutazioni in questo disturbo influenzano la funzione del tRNA mitocondriale, che porta alla rottura del processo globale di intramitocondriale sintesi proteica. Le misurazioni di attività enzimatiche respiratorie nei mitocondri intatti hanno rivelato che più della metà dei pazienti con sindrome di MELAS può avere l’I complessi o complesso I + carenza IV. Un primo rapporto è apparente tra MELAS e carenza del complesso. La sintesi proteica è diminuito può infine portare alla diminuzione osservata dell’attività della catena respiratoria da traduzione ridotto di geni UUG-ricchi come ND6 (componente del complesso). [4]
Inoltre, studi hanno rivelato che il 3243 A → G mutazione produce un grave difetto catena respiratoria combinato in mioblasti, con quasi totale assenza di assemblaggio del complesso I, IV e V, e una leggera diminuzione del complesso assemblato III. Questo difetto di assemblaggio avviene nonostante una modesta riduzione del tasso globale di sintesi delle proteine mitocondriali. Traduzione di alcuni polipeptidi è diminuita, e le prove di aminoacidi misincorporation è notato in altri.
Epidemiologia
Frequenza
Stati Uniti
Nessun stime relative alla prevalenza della mutazione MELAS comuni sono disponibili per la popolazione del Nord America; tuttavia, la sindrome è stata osservata essere meno frequente nei neri.
Internazionale
La prima valutazione della epidemiologia dei disturbi mitocondriali trovato una prevalenza di oltre il 10,2 per 100.000 per la mutazione m.3243A → G nella popolazione finlandese adulta. Se il presupposto è fatto che tutti i parenti materni di primo grado di un portatore di mutazione verificato porto anche la mutazione, prevalenza aumenta a più di 16,3 per 100.000. Questa alta prevalenza suggerisce che i disordini mitocondriali possono costituire una delle maggiori categorie diagnostiche di malattie neurogenetiche tra gli adulti. In Inghilterra del Nord, la prevalenza di questa mutazione nella popolazione adulta è stato determinato in circa 1 ogni 13.000.
Mortalità / morbilità
La malattia progressiva ha una morbilità e mortalità. Il Encefalomiopatia, associata a episodi di ictus seguiti da emiplegia e emianopsia, è grave. Convulsioni focali e generali possono verificarsi in associazione con questi episodi.
Altre anomalie che possono essere osservati sono dilatazione ventricolare, atrofia corticale, e gangli basali calcificazione. Deterioramento mentale di solito progredisce attacchi episodici dopo ripetuti. Anomalie psichiatrici e declino cognitivo (ad esempio, alterazione dello stato mentale, schizofrenia ) possono accompagnare gli episodi di simil-ictus. disturbo bipolare è un’altra anomalia psichiatrica osservata nella sindrome MELAS. disturbi dello spettro autistico (ASD), con o senza ulteriori funzioni neurologiche possono essere prime presentazioni del m 0,3243 A → G mutazione. Miopatia può essere debilitante.L’encefalopatia può progredire verso la demenza; infine, il decorso clinico declina rapidamente, portando a grave disabilità e morte prematura.
Un’altra causa di elevata mortalità è la caratteristica meno comune di coinvolgimento cardiaco, che possono includere cardiomiopatia ipertrofica ,ipertensione e anomalie di conduzione, come blocchi atrio-ventricolare, sindrome del QT lungo , o sindrome di Wolff-Parkinson-White . Sono stati trovati soggetti con sindrome di MELAS aver aumentato crescente rigidità aortica e dell’aorta dimensioni ingrandite suggerendo rimodellamento vascolare. Dissezione aortica radice è stata trovata in un paziente con la sindrome MELAS. [5] Alcuni pazienti possono sviluppare la sindrome di Leigh (cioè, subacuta necrotizzante encefalopatia). I pazienti possono sviluppare insufficienza renale a causa di glomerulosclerosi focale segmentale.
Più raramente, questi pazienti possono presentare gravi disturbi della motilità gastrointestinale e disfunzione endocrina, compreso l’ipotiroidismo el’ipertiroidismo .
Gara
Nessuna predilezione per un particolare gruppo etnico è notato.
Sesso
Nessuna predilezione sessuale è presente.
Età
In molti pazienti con sindrome di MELAS, la presentazione avviene con il primo episodio di ictus, di solito quando un individuo è affinato 4-15 anni. Meno spesso, l’esordio della malattia può avvenire nell’infanzia con tappe dello sviluppo in ritardo e difficoltà di apprendimento. Una presentazione del disturbo è stato segnalato in un neonato di 4 mesi.
Storia
Insorgenza del disturbo può essere miopatica con debolezza, facile affaticamento, e intolleranza all’esercizio.
Miopatia mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) sindrome esordio possono verificarsi nella prima infanzia con una storia di ritardo dello sviluppo e difficoltà di apprendimento. Il ritardo dello sviluppo, difficoltà di apprendimento, o disturbo da deficit di attenzione si trova principalmente in pazienti prima dello sviluppo del primo colpo. Un quadro encefalopatica che è progressiva e porta a demenza può essere presente. I pazienti possono essere apatico. Il livello di funzionamento cognitivo peggiora nel corso del tempo in base al punteggio Karnofsky in pazienti completamente sintomatici.
Ritardo di crescita può essere la funzione di presentare in alcuni pazienti con sindrome MELAS.
Episodi di simil-ictus sono la caratteristica caratteristica di questo disturbo.Inizialmente, gli episodi possono manifestarsi con vomito e mal di testa che può durare diversi giorni. Questi pazienti possono anche sperimentare episodi di crisi epilettiche e anomalie visive seguiti da emiplegia. Tipi di crisi possono essere tonico-cloniche o mioclonica.
Emicrania mal di testa o migrainelike osservati in questi pazienti possono anche riflettere gli episodi di simil-ictus. Pedigree dei pazienti con sindrome MELAS classica identificare molti membri le cui manifestazioni sono solo mal di testa.
I pazienti possono avere lamentele visivi a causa di oftalmoplegia, e possono sperimentare la cecità a causa di atrofia ottica e le difficoltà con visione notturna a causa della retinite pigmentosa.
Alcuni pazienti possono avere perdita di udito, che può accompagnare il diabete. Si può osservare in associazione con il classico disturbo della sindrome MELAS. [8]
Polidipsia e poliuria possono essere i segni che presentano del diabete; diabete sembra essere la manifestazione più comune della sindrome MELAS. Di solito, il diabete di tipo 2 è descritto in individui con sindrome di MELAS, anche se di tipo 1 (precedentemente chiamato diabete insulino-dipendente), può anche essere osservato. Linee guida per la diagnosi e la gestione di tipo sono state stabilite diabete 2. [9]
Palpitazioni e mancanza di respiro possono essere presenti in alcuni pazienti con sindrome MELAS secondaria ad anomalie della conduzione cardiaca, come la sindrome di Wolff-Parkinson-White. I pazienti possono sperimentare mancanza di respiro secondaria a cardiomiopatia, che di solito è di tipo ipertrofico; tuttavia,cardiomiopatia dilatativa è stato anche descritto.
Insorgenza acuta di manifestazioni gastrointestinali (ad esempio, insorgenza acuta di dolore addominale) può riflettere pancreatite , colite ischemica, e ostruzione intestinale. [10]
Intorpidimento, formicolio e dolore alle estremità possono essere manifestazioni di neuropatia periferica.
Disturbi psichiatrici (ad esempio, depressione, disturbo bipolare), sono stati associati con il m.3243 A → G mutazione. La demenza è stata un’altra manifestazione clinica. Inoltre, i disturbi dello spettro autistico (ASD) sono stati associati con il 3243 A → G mutazione.
I pazienti possono sviluppare caratteristiche di ipotiroidismo e ipertiroidismo
Alcuni pazienti possono sviluppare apnea e un’andatura atassica in associazione con le caratteristiche neuroradiologiche della sindrome MELAS.
Oliguria possono essere associati con la sindrome MELAS e può indicare l’insorgenza della sindrome nefrosica .
I pazienti con sindrome di MELAS possono avere coinvolgimento vascolare funzionale. Aortica dissezione radicale è stato riportato in un paziente con la sindrome MELAS.
Esame Fisico
I pazienti con sindrome di MELAS possono presentare ipertensione.
Miopatia presenta con ipotonia e debolezza. Muscoli prossimali tendono ad essere più coinvolti di muscoli distali. Muscolatura è sottile, ei pazienti possono presentare con un volto miopatico.
Episodi di simil-ictus possono presentare convulsioni, anomalie visive, intorpidimento, emiplegia, e l’afasia. [11] episodi può essere seguita da emiplegia transitoria o emianopsia, che dura un paio d’ore a diverse settimane. Altre caratteristiche di esame neurologico possono includere atassia, tremore, mioclono, distonia, disturbi visivi, e la cecità corticale. Alcuni pazienti possono presentare oftalmoplegia e ptosi.
In esame oftalmologico, i pazienti hanno presentato con retinite pigmentosa.
La sordità neurosensoriale è stato segnalato come parte della malattia in circa il 25% dei pazienti con sindrome MELAS.
Cardiomiopatia con segni di insufficienza cardiaca congestizia (CHF) può anche essere osservato dopo un esame fisico. [12]
Manifestazioni cutanee di porpora cutanea, irsutismo, e squamosa, pruriginosa, eritema diffuso con reticolare pigmentazione possono essere osservati nei pazienti con sindrome MELAS.
Bassa statura può essere la prima manifestazione della sindrome MELAS in molti pazienti.
Cause
Sindrome MELAS è stata associata con almeno 6 mutazioni puntiformi differenti, di cui 4 si trovano nello stesso gene, il tRNA Leu (UUR) gene. La mutazione più comune, che si trova nel 80% degli individui con sindrome di MELAS, è una transizione A → G al nucleotide (nt) 3243 nel tRNA Leu (UUR) gene. Un ulteriore 7,5% ha un eteroplasmica mutazione puntiforme T → C a 3271 bp nella coppia nucleotide terminale del gambo anticodone del tRNA Leu (UUR) gene. Inoltre, un fenotipo MELAS è stato osservato associato ad un m.13513G → Una mutazione nel ND5 gene e in carenza di POLG.
Queste mutazioni sono eteroplasmica, che riflette le diverse percentuali di mutato mtDNA presenti in diversi tessuti. Eteroplasmia variabile tra gli individui affetti da sindrome di MELAS riflette segregazione variabile l’ovulo. Mutazioni in tRNA Lys si può aspettare di avere un effetto importante sulla traduzione e la sintesi proteica nei mitocondri. Il mitocondriale umano MELAS disturbo associata tRNA Leu (UUR)mutazione causa la carenza aminoacilazione e un difetto concomitante inizio della traduzione.
Omeostasi del calcio anormale conseguente danno neuronale è stato suggerito come un altro meccanismo che contribuisce al coinvolgimento CNS osservata nella sindrome MELAS.
I pazienti con sindrome MELAS sono stati trovati per avere una marcata diminuzione dell’attività del complesso I. Gli effetti principali osservati secondaria a nt 3243 e 3271 nt mutazioni sono state una riduzione della sintesi proteica e l’attività del complesso I. Questi effetti sono stati dimostrati attraverso cibridi che studiano in cui le linee cellulari umane senza mtDNA si fondono con mitocondri esogeni contenenti 0-100% della mutazione m.3243 comune. Cybrids con più del 95% di DNA mutante era diminuita velocità di sintesi delle proteine mitocondriali, portando a difetti della catena respiratoria.
Procedere alla Diagnosi differenziale
Diagnosi differenziale
· Sindrome da anticorpi antifosfolipidi
· Blocco atrioventricolare, Second Degree
· Blocco atrioventricolare, Terzo Grado, acquisita
· Lunga catena acil CoA deidrogenasi, deficit di
· A catena media acil-CoA deidrogenasi
· Mitocondriale DNA polimerasi carenza (POLG)
· Disturbo dell’Umore: disturbo bipolare
· Disturbo dell’Umore: Depressione
· Oliguria
· Pancreatite e pseudocisti pancreatica
· Tachicardia sopraventricolare, sindrome di Wolff-Parkinson-White
Studi di laboratorio
I seguenti studi sono indicati nei pazienti con miopatia mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) sindrome:
· Acido siero lattico, acido piruvico siero, liquido cerebrospinale (CSF) di acido lattico e acido piruvico CSF
o L’acidosi lattica è una caratteristica importante della sindrome MELAS.Vedere l’immagine qui sotto per la classificazione fisiopatologico di acidosi lattica.
classificazione fisiopatologico di acidosi lattica.
o In generale, acidosi lattica non porta ad acidosi metabolica sistemica, e può essere assente nei pazienti con notevole coinvolgimento del SNC.
o In alcuni individui con sindrome di MELAS, livelli di acido lattico possono essere normali nel sangue, ma elevata in CSF.
o In difetti della catena respiratoria, il rapporto tra lattato e piruvato è alta.
· Livelli di creatina chinasi sierici
o I livelli sierici di creatina chinasi sono leggermente ad aumentare moderatamente in alcuni pazienti con sindrome MELAS.
o Livelli tendono ad aumentare durante e immediatamente dopo episodi.
· Respiratori attività degli enzimi della catena nel muscolo scheletrico
o Se una biopsia muscolare viene eseguita per perseguire una valutazione diagnostica, quindi verificare le attività degli enzimi della catena respiratoria.
o I pazienti con sindrome di MELAS sono stati trovati ad avere segnato la carenza di attività complesso della catena respiratoria I.
o Alcuni pazienti con la malattia hanno un deficit combinato del complesso I e IV complessa.
· Mitocondriale analisi della mutazione del DNA sul sangue, muscolo scheletrico, follicoli dei capelli, mucosa orale, e sedimento urinario
o Individui con più gravi manifestazioni cliniche della sindrome MELAS hanno generalmente superiore al 80% mtDNA mutante nei tessuti stabili come muscolare.
o In cellule in rapida divisione, come i componenti delle linee ematopoietiche, la m.3243 A → G mutazione può separare a livelli estremamente bassi, rendendo la diagnosi genetica dal sangue difficile.
o La percentuale della mutazione diminuisce progressivamente in DNA isolato dal sangue. Il carico mutante isolata dal sangue non è né utile per la prognosi né per la valutazione funzionale.
o Sedimento urinario, seguita da fibroblasti della pelle e mucosa orale, sono i tessuti accessibili della scelta, perché sono di facile accesso e il carico di mutazione è maggiore di quella riscontrata nel sangue.
o Se la diagnosi si sospetta ancora dopo normali risultati delle analisi di mutazione del mtDNA in questi tessuti, una biopsia muscolare scheletrico è necessario per confermare o escludere la presenza della mutazione.
Studi di imaging
I seguenti studi di imaging possono essere presentate:
· TAC o risonanza magnetica del cervello
o TAC o risonanza magnetica del cervello a seguito di un episodio di ictus rivela un lucency coerente con infarto.
o Più tardi, atrofia cerebrale e calcificazioni possono essere osservati in studi di brain imaging.
o I pazienti con sindrome di MELAS che hanno una presentazione simile alla sindrome di Leigh possono avere calcificazioni nei gangli basali.
· Tomografia ad emissione di positroni (PET) studi
o Gli studi PET possono rivelare un tasso metabolico cerebrale ridotta per l’ossigeno.
o Aumento flusso sanguigno cerebrale nelle regioni corticali può osservare.
o PET può dimostrare di mantenimento del tasso metabolico cerebrale per il glucosio.
· Singolo fotone studi CT emissione
o Emissione di singoli fotoni tomografia computerizzata (SPECT) studi possono accertare colpi in individui con sindrome MELAS con un tracciante, N -isopropyl-p- [123-I] -iodoamphetamine.
o Il tracciante accumula nella regione parietooccipital, e può delineare l’estensione della lesione. Studi SPECT vengono utilizzati per monitorare l’evoluzione della malattia.
· Spettroscopia di risonanza magnetica protonica ( 1 H-MRS): questo viene utilizzato per identificare anomalie metaboliche, tra cui il rapporto lattato-to-creatina sia muscolo o cervello e il sistema nervoso centrale è diminuito Nrapporto -acetylaspartate-to-creatina nelle regioni di ictus. Con questa tecnica, regioni elevati di lattato sono stati rilevati mentre i livelli sierici sono normali.
· Ecocardiografia: Questo è utile per valutare per cardiomiopatia ipertrofica e dilatativa e le dimensioni della radice aortica; tuttavia, cardiomiopatia non è una caratteristica comune nelle persone con sindrome MELAS.
Altri test
EEG di solito sono anormali. Scariche epilettiformi picco sono di solito presenti.
ECG viene utilizzato per cercare anomalie di conduzione con aritmie ventricolari.ECG può identificare coinvolgimento presintomatica cardiaco, sindromi pre-eccitazione, e blocco di conduzione cardiaca.
Procedure
Considerare l’esecuzione di una biopsia muscolare, se la sindrome MELAS si sospetta e se l’analisi di mutazione del mtDNA nel sangue e altri tessuti accessibili fornisce risultati banali. In rapida divisione linee di cellule, le mutazioni possono separare a livelli bassi, rendendo la diagnosi genetica dal sangue difficile.
I risultati istologici
In biopsie muscolari colorate con ematossilina eosina, la variazione si osserva nelle misure di tipo 1 e di tipo 2 in fibra, che rappresenta alterazioni miopatiche.
Fibre rosse sfilacciate sono il segno distintivo della sindrome MELAS. Le fibre rosse sfilacciate colorano rosso brillante con corpi citoplasmatici occasionali con colorazione tricromica. Fibre rosse sfilacciate solito macchia positiva con macchia citocromo ossidasi.
Colorazione con acido periodico di Schiff, nicotinamide adenina dinucleotide (NADH) deidrogenasi tetrazolio reduttasi, o per succinico deidrogenasi dimostra una maggiore attività subsarcolemmale. Questa proliferazione mitocondriale è stato osservato anche nei vasi sanguigni e viene determinato utilizzando una macchia per la succinato deidrogenasi.
La microscopia elettronica mostra un aumento del numero e le dimensioni dei mitocondri, alcuni con corpi paracristalline.
Cura Medica
La valutazione per la sindrome (MELAS) miopatia mitocondriale, encefalopatia, acidosi lattica, ed ictus può essere eseguita su base ambulatoriale, se il paziente è stabile. La valutazione può essere costituita da nella determinazione dei livelli di lattato nel siero e piruvato sierico, studi sulle mutazioni del mtDNA sul sangue, e studi di brain imaging (ad esempio, la scansione testa CT, MRI del cervello, spettroscopia di risonanza magnetica [ 1 H-MRS]) protonica del cervello. Può essere eseguita come procedura elettiva la biopsia muscolare per gli enzimi mitocondriali e l’analisi di mutazione del DNA per cui il paziente è ricoverato in ospedale.
In casi di scompenso acuto, effettuare studi pazienti ricoverati in fase acuta e in seguito la stabilizzazione del paziente.
Sono disponibili varie misure di sostegno, anche se nessuno studio controllato ha dimostrato efficacia. I benefici a lungo termine di manipolazioni dietetiche sono sconosciuti. I miglioramenti in alcuni pazienti possono essere correlati a una migliore stato nutrizionale e idratazione.
Sono stati utilizzati i seguenti farmaci:
· Il trattamento con coenzima CoQ10 è stato utile in alcuni pazienti con sindrome MELAS. Nessun effetto negativo sono stati riportati dalla sua amministrazione.
· Menadione (vitamina K-3), phylloquinone (vitamina K-1), e ascorbato sono stati utilizzati per donare elettroni citocromo c . Idebenone è stato utilizzato anche per il trattamento di questa condizione, e sono stati segnalati miglioramenti nella alterazioni cliniche e metaboliche.
· Riboflavina è stato segnalato per migliorare la funzione di un paziente con carenza del complesso e la m.3250 T → C mutazione. Nicotinamide è stata utilizzata perché complesso accetta elettroni da nicotinamide adenina dinucleotide (NADH) e infine trasferisce elettroni CoQ10.
· Dicloroacetato è un altro composto usato con questi agenti, poiché i livelli di lattato si abbassano nel plasma e nel liquido cerebrospinale (CSF); pazienti riferito possono rispondere in modo favorevole. La neuropatia sensoriale può causare dopo un uso prolungato di questo farmaco.
· Succinato di sodio è stato utilizzato, e un paziente con la sindrome MELAS riferito avuto meno episodi di simil-ictus con il suo uso; tuttavia, succinato di sodio non è lo standard di cura. Ulteriori indagini è necessario.
· Creatina monoidrato è stato utilizzato anche, e un aumento della forza muscolare in attività anaerobiche e aerobiche ad alta intensità è stata riportata.
· La somministrazione di L-arginina nei periodi acuti e interictali può rappresentare una nuova potenziale terapia per questa sindrome per ridurre il danno cerebrale dovuto alla vasodilatazione alterata in arterie intracerebrali causa ossido nitrico esaurimento.
Consulti
I seguenti consultazioni possono essere presentate:
· Genetista
· Neurologo (per valutare il paziente per gli episodi ictus)
· Cardiologo (per la valutazione di cardiomiopatia, aritmie e ipertensione)
· Nefrologo (da valutare per l’insorgenza della sindrome nefrosica)
· Oculista (di valutare per la retinopatia pigmentosa)
· Endocrinologo (valutare per disfunzioni endocrine come il diabete mellito, ipotiroidismo, ipertiroidismo e ipoparatiroidismo)
· Psichiatra (per valutare per i disturbi affettivi)
· Neuropsicologo (valutare per disturbi dello spettro autistico [ASD])
Dieta
L’effetto della manipolazione alimentare non è completamente noto, e l’efficacia di integratori alimentari non è provata. Aciduria dicarbossilici e compromissione secondaria di acidi grassi a catena lunga ossidazione (LCFAO) possono verificarsi nei disturbi mitocondriali. Miglioramento osservato in molti pazienti è probabilmente legato a una migliore alimentazione.
Attività
Nei pazienti con miopatie mitocondriali, formazione tapis roulant moderata può comportare il miglioramento della capacità aerobica e un calo dei livelli di lattato di riposo e postexercise lattato. Allenamento concentrico può anche giocare un ruolo importante perché dopo un breve periodo di esercizio concentrica formazione di un notevole incremento riferito avviene nel rapporto di tipo-to-mutante mtDNA selvatici e della percentuale di fibre muscolari con normale attività della catena respiratoria.
Riassunto Farmaci
Per gli individui con miopatia mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) sindrome e per quelli con altri disturbi della fosforilazione ossidativa (OXPHOS), terapie metabolici vengono somministrati per aumentare la produzione di adenosina trifosfato (ATP) e per rallentare o arrestare il deterioramento di questa condizione e di altri encefalomiopatie mitocondriali. Terapie metaboliche utilizzate per la gestione della sindrome MELAS comprendono carnitina, CoQ10, fillochinone, menadione, ascorbato (vale a dire, l’acido ascorbico), riboflavina, nicotinamide, creatina monoidrato, idebenone, succinato, e dicloroacetato. Tuttavia, la valutazione dell’efficacia di questi composti è lungi dall’essere completa, ed efficacia si ritiene essere limitata ai casi individuali.
Il trattamento con CoQ10 è stato utile in alcuni pazienti con sindrome MELAS. Nessun effetto negativo sono stati riportati dalla sua amministrazione. Menadione (vitamina K-3), phylloquinone (vitamina K-1), e ascorbato sono stati utilizzati per donare elettroni citocromo c . Idebenone è stato utilizzato anche per il trattamento di questa condizione, e sono stati segnalati miglioramenti nella alterazioni cliniche e metaboliche. Riboflavina è stato segnalato per migliorare la funzione di un paziente con carenza del complesso e la m.3250 T → C mutazione. Nicotinamide è stata utilizzata perché complesso accetta elettroni da nicotinamide adenina dinucleotide (NADH) e infine trasferisce elettroni Q10. Dicloroacetato è un altro composto usato con questi agenti, perché i livelli di lattato si abbassano nel plasma e nel liquido cerebrospinale (CSF). I pazienti riferito possono rispondere in modo favorevole.
Un paziente con la sindrome MELAS riferito ha avuto meno episodi di simil-ictus con l’uso di succinato di sodio; tuttavia, succinato di sodio non è lo standard di cura, e sono necessarie ulteriori indagini. Un aumento della forza muscolare in attività anaerobica e aerobica ad alta intensità è stata riportata con la somministrazione di creatina monoidrato.
La somministrazione di Arginina nei periodi acuti e interctritici degli episodi di simil-ictus della sindrome MELAS, può rappresentare una nuova potenziale terapia per ridurre i danni al cervello a causa della disfunzione mitocondriale, ed è una delle terapie più promettenti ad oggi. Sulla base l’ipotesi che gli episodi di simil-ictus nella sindrome MELAS sono attivati da vasodilatazione alterata nelle arterie cerebrali dovute alla diminuzione dei livelli di circolante NO, elevazione di arginina e livelli di NO può migliorare questo effetto. Inoltre, L-arginina può modulare l’eccitazione da neurotrasmettitori a terminazioni nervose e tali effetti potrebbe contribuire ad alleviare i sintomi simil-ictus nella sindrome MELAS. I pazienti con MELAS possono avere meno probabilità di avere episodi di ictus, migliorando la loro funzione endoteliale con la supplementazione orale di L-arginina.
Vitamine e integratori alimentari
Riassunto Class
Le vitamine sono sostanze organiche del corpo richiede in piccole quantità per vari processi metabolici. Le vitamine possono essere sintetizzati in piccole o insufficienti quantità nel corpo o no sintetizzati a tutti, richiedendo così l’integrazione. Alcuni case report che utilizzano integratori alimentari hanno riportato un miglioramento dei sintomi del paziente.
Visualizza informazioni complete droga
Può essere utile per il trattamento / prevenzione di episodi di ictus nella sindrome MELAS. Gli episodi di simil-ictus nella sindrome MELAS può essere innescato da vasodilatazione alterata nelle arterie cerebrali dovute alla diminuzione dei livelli circolanti di NO; quindi, elevazione di arginina e aumentata sintesi di NO può migliorare questo effetto.
Aumenta la produzione di ornitina, che facilita l’incorporazione di azoto rifiuti nella formazione di citrullina e argininosuccinato. Fornisce 1 mol di urea più 1 mol ornitina per mole di arginina quando spaccati da arginase.
Visualizza informazioni complete droga
Un derivato di aminoacido, sintetizzato da metionina e lisina, richiesto nel metabolismo energetico. Può promuovere l’escrezione degli acidi grassi in eccesso nei pazienti con difetti nel metabolismo degli acidi grassi o acidopathies organici specifici che causano esteri acil CoA di bioaccumulo.
In carenza di carnitina secondaria associata alla sindrome MELAS, carnitina può ripristinare generazione di liberi CoA ed evitare carnitina esaurimento. Se si verifica la sindrome MELAS associata LCFAO difetti, uso della carnitina è discutibile perché può aumentare la formazione di acilcarnitine a catena lunga, che possono causare aritmogenesi ventricolare.
Visualizza informazioni complete droga
Un chinone liposolubile, la cui funzione è trasferimento di elettroni dal complesso I al complesso III. Sembra stabilizzarsi complessi OXPHOS situati in membrana mitocondriale interna; può anche agire come potente antiossidante per i radicali liberi. È stato osservato Miglioramento della debolezza muscolare e una diminuzione di lattato sierico.
Idebenone (Avan)
I dati sono limitati; Tuttavia, si ritiene che permettono di migliorare il metabolismo cerebrale e migliorare la funzione di sistema elettronico di trasferimento dei mitocondri cerebrali. Inoltre inibisce la perossidazione lipidica della membrana mitocondriale, quindi, aumentando l’attività respiratoria mitocondriale.
È stato usato per il trattamento di pazienti affetti da sindrome MELAS sulla base di effetti fisiologici proposti come antiossidante, effetto presunta sulle svalutazioni di breve termine e memoria a lungo termine, e la somiglianza strutturale CoQ10. Non approvato per l’uso in pazienti negli Stati Uniti; Tuttavia, è stato utilizzato in Giappone. Miglioramento anomalie cliniche e metaboliche si osserva nei pazienti con sindrome MELAS. Non sono noti effetti avversi.
Visualizza informazioni complete droga
Dopo la conversione di flavina monofosfato e flavina adenina dinucleotide, funziona come cofattore per il trasporto degli elettroni in complesso I, complesso II, e il trasferimento di elettroni flavoproteina. Secondo quanto riferito di beneficio in caso di carenza del complesso e MELAS.
Visualizza informazioni complete droga
Può essere utile nei pazienti individuali come antiossidante.
Menadione (vitamina K-3)
È stato riportato aneddoticamente per migliorare il metabolismo del fosfato cellulare; migliora tasso di riduzione fumarato permettendo il trasferimento di elettroni da S3 gruppo di zolfo ferro del complesso II; sembra migliorare il trasferimento di elettroni dopo complesso I inibizione da rotenone. Anche se il passaggio attraverso la placenta è povero, somministrare con cautela nei pazienti in stato di gravidanza con sindrome MELAS vicino al termine, perché emolisi e iperbilirubinemia riferito hanno colpito i neonati.
Visualizza informazioni complete droga
Può avere effetti benefici nei pazienti con MELAS e altri disturbi mitocondriali;effetto può essere correlato ad un aumento della creatina intracellulare e / o contenuti fosfocreatina, che possono essere coinvolte nel mantenimento ATP cellulare e nella stabilizzazione della permeabilità transizione pore con conseguente morte neuronale per apoptosi. L’assunzione di creatina può aumentare la forza muscolare nei pazienti con sindrome MELAS (osservati in un paziente con la sindrome MELAS arruolati in uno studio). Effetto citotossico potenziale di somministrazione a lungo termine.
Dicloroacetato sodio (ceresina)
Attualmente un farmaco orfano negli Stati Uniti. Un composto creduto per attivare il complesso della piruvato deidrogenasi inibendo chinasi inattivante. Questo diminuisce la produzione di lattato e promuove piruvato ossidazione. Usato per abbassare i livelli di lattato sia nel plasma e CSF. Al momento è disponibile solo in protocolli di ricerca. Effetto primario è quello di stimolare la funzione di PDH inibendo chinasi che inattiva PDH. Inoltre può stimolare glycolytic enzima fosfofruttochinasi sopprimendo inibitore allosterico (citrato) e aumentando i livelli di attivatore (fruttosio 2,6 biphosphate) per migliorare l’ossidazione del lattato nel fegato.
Ulteriore assistenza ospedaliera
Ammetta di scompenso o segni di chetoacidosi diabetica metabolica. Il diabete sembra essere la manifestazione più comune di miopatia mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) sindrome. Ammetta per la gestione medica di episodi di simil-ictus e crisi epilettiche. Ammettere di segni di aritmia cardiaca (sindrome di Wolff-Parkinson-White), ipertensione, imminente dissezione radice aortica, o insufficienza cardiaca congestizia (CHF) associata a cardiomiopatia ipertrofica o dilatativa. Ammettere di segni di sindrome nefrosica, che possono presentare in associazione con glomerulosclerosi focale segmentale.Ammettere se un segno di addome acuto è presente; addome acuto può essere un’indicazione di pancreatite. [13]
Ulteriori cure ambulatoriali
Monitorare attentamente l’andamento della encefalomiopatia e sequele. Test sviluppo neurologico è appropriato perché progressivo deterioramento intellettuale segue episodi ictus della sindrome MELAS. Valutazione neuropsicologica è appropriato per la presenza di disturbi dello spettro autistico (ASD).
Monitorare le curve di crescita, perché le malattie mitocondriali, come la sindrome MELAS sono associati con bassa statura e ritardo di crescita.
Fare riferimento al paziente di un oftalmologo per monitorare degenerazione pigmentosa della retina, che può essere simile a quello osservato nei pazienti con neuropatia, atassia e sindrome retinite pigmentosa. Seguire attentamente le indicazioni (ad esempio, oftalmoplegia, ptosi).
Monitorare attentamente le persone con sindrome di MELAS per la perdita dell’udito con una valutazione dell’udito, tra cui prodotti distorsione otoemissioni acustiche e del tronco encefalico evocati uditivi risposte. Monitorare attentamente i pazienti per cardiomiopatia e misurare Z-score per il diametro della radice aortica con ecocardiografia. Richiedi un ECG come uno studio di riferimento per monitorare i difetti di conduzione, anche se i pazienti sono asintomatici. Monitorare attentamente i pazienti per il diabete di tipo 2, ipotiroidismo, ipertiroidismo, e disfunzione paratiroidea. Monitorare attentamente i pazienti per la persistenza di acidosi lattica.
Spettroscopia di risonanza magnetica positroni ( 1 H-MRS) del cervello può essere usato per monitorare potenziale efficacia terapeutica se aumentata permeabilità della barriera emato-encefalica è una preoccupazione.
Farmaci Ospedalieri e Ambulatoriali
Farmaci includono i seguenti:
· Composti che possono aumentare la produzione o il trasferimento di elettroni ATP (ad esempio, ascorbato, riboflavina, CoQ10, vitamine K-1 e K-3, nicotinamide, creatina monoidrato)
· Composti che possono essere utilizzati per prevenire una possibile carenza di carnitina secondaria o disfunzione secondaria di ossidazione degli acidi grassi (ad esempio, carnitina)
· Composti che possono essere utilizzati per prevenire o migliorare la progressione di episodi ictus (ad esempio, L-arginina): L-arginina potrebbe modulare il metabolismo energetico mitocondriale inibendo glutammato assorbimento nei mitocondri e diminuendo neurotossicità associata a disfunzione mitocondriale ossido nitrico-mediata.
· Composti che possono essere usati per trattare l’acidosi lattica (ad esempio, dicloroacetato)
o Dicloroacetato stimola la funzione piruvato deidrogenasi inibendo piruvato chinasi deidrogenasi, l’enzima che fosforila normalmente e inattiva piruvato deidrogenasi. Pertanto, in condizioni che provocano l’accumulo di lattato e alanina, attivazione di piruvato deidrogenasi diminuisce il rilascio di questi composti dai tessuti periferici e migliora il loro metabolismo ossidativo dal fegato.
o Questo farmaco è stato utilizzato per il trattamento di acidosi lattica in pazienti adulti e pediatrici. Aneddotica riporta dettagliatamente il successo del trattamento nei pazienti con sindrome MELAS. Dicloroacetato è stato somministrato per via orale a dosi di 12,5-100 mg / kg / d. Questo farmaco è disponibile solo in protocolli di ricerca negli Stati Uniti.
Se sequestri si sono sviluppati come parte della condizione, non utilizzare l’acido valproico come anticonvulsivante, dal momento che gli episodi di pancreatite a seguito di somministrazione valproato sono verificati e acido valproico è stato associato a tossicità mitocondriale.
Utilizzare fenobarbital con cautela, perché il farmaco ha dimostrato un’inibizione della catena respiratoria in vitro.
Trasferimento
Trasferire in un centro di cura terziario può essere richiesto di coordinare meglio la valutazione diagnostica per includere i seguenti:
· La biopsia muscolare
· Valutazione per difetti enzimatici mitocondriali
· Analisi di mutazione del mtDNA
Se la diagnosi è già nota e il paziente è stato stabilizzato, il trasferimento può essere richiesta per una migliore gestione delle complicanze, come la seguente:
· Pancreatite
· Aritmie cardiache
· Cardiomiopatia
· Chetoacidosi
· Episodi di simil-ictus
Deterrenza / Prevenzione
Se le condizioni, come la cardiomiopatia sono presenti, limitare l’esercizio. Anche se gli effetti a lungo termine di manipolazioni dietetiche sono sconosciuti, garantire un buono stato nutrizionale, una buona idratazione, ed evitare il digiuno come parte di un piano di sostegno. Un grado lieve di attività aerobica può portare ad un miglioramento della capacità aerobica. Limitare intenso esercizio fisico a causa della possibile complicanza di rabdomiolisi.
Informazioni sulla efficacia terapeutica dei composti riportati utilizzati come integratori nutrizionali sono limitati; tuttavia, la maggior parte non ha effetti avversi gravi. Gli integratori alimentari possono aiutare a prevenire un ulteriore deterioramento in alcuni individui; Tuttavia, ulteriori ricerche è giustificato.
Complicazioni
Le complicazioni sono i seguenti:
· Ritardo di crescita e bassa statura
· Progressivo deterioramento intellettuale e il declino che alla fine può portare a demenza
· Psicosi con la depressione, la schizofrenia, disturbo bipolare
· Disturbi dello spettro autistico (ASD)
· Ipoacusia neurosensoriale
· Endocrine erettile con ipogonadismo, diabete, ipoparatiroidismo, ipotiroidismo, ipertiroidismo e
· CHF da cardiomiopatia e morte improvvisa da difetti di conduzione
· Difficoltà visive legate alla degenerazione pigmentaria della retina o cecità corticale come uno dei postumi di atrofia corticale progressiva ed episodi di simil-ictus
· Insufficienza renale allo stadio terminale come complicanza di glomerulosclerosi focale segmentale
· Insufficienza renale acuta secondaria a rabdomiolisi
· GI disfunzione secondaria a pseudoobstruction intestinale o pancreatite
· Aortica dissezione root (riportato in un gruppo di affini, richiede ulteriori studi per valutare la prevalenza)
Prognosi
Sindrome MELAS ampiamente varia nella presentazione; tuttavia, i pazienti in generale tendono ad avere una prognosi sfavorevole e l’esito. Il Encefalomiopatia tende ad essere grave e progressiva demenza. Il paziente con la sindrome MELAS può finire in uno stato di cachessia. Attualmente, esistono terapie hanno dimostrato efficacia.
Diabete e Sordità, Maternamente Ereditati- (Maternally Inherited Diabetes mellitus and Deafness MIDD)–Sindrome Ballinger-Wallace–Diabete Mellito, di Tipo II, con La Sordità–Diabete Mellito non Insulino-Dipendente Con La Sordità–NIDDM con la Sordità
Diabete e Sordità, Maternamente Ereditati( MIDD)- pag.57
Penetranza e l’età di esordio pag.57
Sintomi pag.57
Effetto della mutazione su tRNALeu (UUR) pag.58
Caratteristiche metaboliche del diabete in MIDD pag.58
Caratteristiche metaboliche della sordità in MIDD pag.58
Il diabete mellito e sordità (DAD) o ereditarietà materna diabete e sordità (MIDD) è un sottotipo di diabete che è causato da una mutazione puntiforme in posizione 3243 in umano DNA mitocondriale , che consiste in un genoma circolare. Questo influenza il tRNALeu gene codificante. [1][2] A causa del fatto che il DNA mitocondriale si trova solo in oociti e non è presente in spermatozoi , questa malattiaè ereditato da soli familiari materni. [1] Come indicato dal nome , MIDD è caratterizzata da diabete e sordità neurosensoriale . [1]
Penetranza ed età di esordio
MIDD rappresenta l’1% dei pazienti con diabete . Oltre l’85% delle persone che portano la mutazione in DNA mitocondriale alla posizione 3243 sintomi presenti di diabete. L’età media in cui i pazienti MIDD sono tipicamente diagnosticati è 37 anni, ma è stato visto di spaziare ovunque tra 11 anni per 68 anni. Di questi pazienti diabetici che trasportano il DNA mitocondriale mutazione alla posizione 3243, l’esperienza il 75% di perdita dell’udito neurosensoriale . [1] In questi casi, la perdita dell’udito viene solitamente visualizzata prima della comparsa del diabete ed è caratterizzato da una diminuzione della percezione di frequenze di tono alto.[3] La perdita dell’udito associata al diabete è in genere più comune e più rapidamente calo negli uomini che nelle donne. [4]
Sintomi
Come suggerito dal nome, pazienti Midd sono soggetti a perdita di udito neurosensoriale . [1] Questo inizia con una riduzione dellapercezione di frequenze superiore a circa 5 Hz che diminuisce progressivamente, nel corso degli anni, alla perdita uditiva severa in tutte le frequenze . [1] Il diabete che accompagna la perdita dell’udito può essere simile al diabete di tipo 1 o diabete di tipo 2 ;tuttavia, diabete di tipo 1, come è la forma più comune dei due. MIDD è stato anche associato a una serie di altre questioni, tra cuirenale disfunzione, gastrointestinali problemi, e cardiomiopatia . [3]
Effetto della mutazione su tRNALeu (UUR)
I mitocondri hanno il loro genoma circolare che contiene 37 geni , di cui 22 codice per tRNA . [5] Questi tRNA svolgono un ruolo essenziale nella sintesi proteica trasportando aminoacidi al ribosoma . [1] MIDD è causato da un A alla sostituzione G nel DNA mitocondriale alla posizione 3243, che codifica tRNALeu (UUR). [1] Questa mutazione è in genere in eteroplasmica forma. Una mutazione in questo gene (A3243G) provoca la conformazione nativa da destabilizzato, nonché dimerizzazione nel tRNALeu (UUR). L’ uridina al anticodone prima posizione del tRNALeu (UUR) è normalmente post trascrizionalmente modificata per garantire il corretto codone riconoscimento. Tale modifica è noto come taurina modifica, che viene diminuito a seguito della struttura improprio del tRNALeu (UUR). [6] errato tRNALeu (UUR) Struttura traduce anche in una riduzione aminoacilazione . [5]La mutazione ha anche dimostrato di risultare in una minore funzione del tRNA e quindi la sintesi proteica . [7]
Caratteristiche metaboliche del diabete in MIDD
La mutazione A3243G nel DNA mitocondriale può essere presente in qualsiasi tessuto, tuttavia, è più comunemente presenti neitessuti con inferiori replica tassi come muscolo . [3] La presenza di questa mutazione può portare a una diminuzione del consumo di ossigeno a causa della ridotta funzionalità della catena respiratoria e una diminuzione della fosforilazione ossidativa . [8] In alcuni pazienti, questa riduzione in funzione della catena respiratoria è suggerito per essere causato da quantità bilanciate diproteine che sono codificati dal DNA mitocondriale , a causa della presenza del A3243G mutazione. [3] Tuttavia, in altri pazienti, la stessa quantità di proteine mitocondriali sono generati, ma la loro stabilità è compromessa a causa della incorporazione improprio di amminoacidi alle UUR codoni delle mitocondrialimRNA . Questo è un risultato del tRNALeu mutato (UUR) con la sua funzione diminuita sintesi proteica . [8] Una diminuzione in funzione della catena respiratoria a seguito di un DNA mitocondriale mutazione può determinare una diminuzione di ATP produzione. Questa diminuzione ATP potrebbe avere effetti negativi su altri processi nel corpo. Un tale processo è l’insulina secrezione da pancreatiche beta-cellule . [3] In pancreatiche beta-cellule , precisi livelli di ATP /ADP regolano l’apertura e la chiusura del canale KATP, che controlla la secrezione di insulina . Quando mutazioni nei mitocondriperturbare il ATP / ADP rapporto, questo canale non può funzionare correttamente e questo può provocare nel paziente essendo carenti di insulina . [3] Poiché l’età di insorgenza è più tardi nella vita di un paziente, è stato suggerito che l’età gioca un ruolo nel contribuire, insieme alla ridotta ATP / ADP rapporto, il lento deterioramento della funzione delle cellule B . [3]
Caratteristiche metaboliche della sordità in MIDD
La perdita di udito , come causata dal 3243 DNA mitocondriale mutazione, è visto in forma di progressiva cocleare erettile.Sebbene il meccanismo con cui la mutazione nella tRNALeu (UUR) causa questa disfunzione della coclea è ancora sotto investivation, è stato ipotizzato che coinvolge le pompe ioniche necessari per il suono trasduzione . [3] Come la mutazione nel tRNALeu (UUR ) porta ad importi non bilanciati o instabili catena respiratoriaenzimi , la respirazione e la fosforilazione ossidativasono ridotti, portando a livelli più bassi di ATP . [3][8] Naturalmente, gli organi più metabolicamente attivi nel paziente saranno interessati da questo ATP carenza. Incluso in questi organi metabolicamente attivi è il coclearestria vascularis . [1] Il vascularis stria e le cellule ciliate , entrambi essenziali al suono trasduzione, fare uso di pompe ioniche per regolare la concentrazione di ioni tra cui K +, Na +, e Ca2 + utilizzando ATP . Senza adeguati livelli di ATP , questi gradienti di concentrazione non vengono mantenuti e questo può portare a morte cellulare in entrambe le vascularis stria e le cellule dei capelli , causando la perdita dell’udito. [9]
Tabella 1: organi metabolicamente attivo che possono essere interessati dalla 3243A> G mutazione puntiforme mitocondriale e la complicazione associati: [1]
|
Organo interessato |
Complicanza associata |
|
Ear ( coclea ) |
|
|
Cervello ( ipotalamo ) |
Bassa statura |
|
Occhio |
Maculare modello distrofia |
|
Cuore |
|
|
Rene |
|
|
Intestino |
Malassorbimento o costipazione |
|
Cervello |
Strokes, atrofia del cervelletto o cervello |
|
Muscolo |
Vedi anche
· diabete
· sordità
LA CLASSIFICAZIONE DELLE MALATTIE MITOCONDRIALI
L’identificazione di mutazioni del mtDNA ha fornito le basi per l’attuale classificazione dei disordini mitocondriali.
Un primo gruppo di malattie è caratterizzato dalla presenza di mutazioni del mtDNA, ad insorgenza sporadica, o a trasmissione materna. Un secondo gruppo è causato da mutazioni in geni nucleari che fanno parte o controllano la fosforilazione ossidativa (OXPHOS). Queste malattie sono spesso classificate sulla base delle sole alterazioni biochimiche rilevate dall’analisi dei tessuti affetti (soprattutto muscolo scheletrico), perché i geni responsabili ancora non si conoscono, anche se molti progressi sono stati recentemente compiuti in questo campo.
1. Mutazioni del mtDNA
A seconda delle caratteristiche molecolari e genetiche delle mutazioni del mtDNA, questo gruppo di difetti comprende sindromi dovute a riarrangiamenti su larga scala del mtDNA, o a mutazioni puntiformi del mtDNA.
Malattie da mutazioni di geni mitocondriali (mtDNA)*
|
Riarrangiamenti del DNA mitocondriale (delezioni)** |
|
Sindrome di Kearns-Sayre (KSS) |
|
Oftalmoplegia Esterna Progressiva (PEO) |
|
Sindrome di Pearson (anemia sideroblastica e malassorbimento connatali) |
|
Mutazioni puntiformi* |
|
Neuropatia ottica ereditaria di Leber (LHON) |
|
Sindrome di NARP (neuropatia, atassia, retinite pigmentosa) |
|
Sindrome MILS (Sindrome di Leigh ereditata per via matrilineare) |
|
Encefalopatia mitocondriale con acidosi lattica e strokes (MELAS) |
|
Mioclono-epilessia con fibre “ragged-red”(MERRF) |
|
Miopatia e cardiomiopatia (MIMYCA) |
* eredità matrilineare
** forme quasi sempre sporadiche
Alterazioni qualitative del mtDNA
Si può trattare di delezioni parziali del mtDNA o, più raramente, di duplicazioni parziali. Entrambi i tipi di mutazione sono eteroplasmici, dato che coesistono sempre con una quota di mtDNA normale. Queste alterazioni grossolane del mtDNA sono quasi invariabilmente associate con tre principali presentazioni cliniche: la sindrome di Kearns-Sayre, l’Oftalmoplegia Esterna Progressiva, e la sindrome di Pearson.
La s. di Kearns-Sayre (KSS) è una grave malattia ad insorgenza sporadica caratterizzata dalla triade: 1) Oftalmoplegia Esterna Progressiva (PEO) con ptosi (abbassamento) palpebrale bilaterale; 2) Retinopatia Pigmentaria; 3) insorgenza prima dei 20 anni. Segni aggiuntivi frequenti sono l’incoordinazione motoria (atassia) di origine cerebellare, il deterioramento mentale, la sordità, e alterazioni del ritmo cardiaco. Vi è spesso ritardo della crescita.
La Oftalmoplegia Esterna Progressiva (PEO) è anch’essa sporadica e caratterizzata dall’insorgenza in età adulta di ptosi bilaterale e paralisi dei movimenti oculari, spesso associate a debolezza dei muscoli delle spalle e del bacino di grado variabile.
La sindrome di Pearson è una rara malattia sporadica della primissima infanzia caratterizzata da anemia sideroblastica, pancitopenia ed insufficienza del pancreas esocrino con malassorbimento intestinale. Nei bambini che sopravvivono oltre i primi anni si assiste ad un progressivo miglioramento della situazione ematologica e gastrointestinale, mentre insorgono i segni caratteristici della sindrome di Kearns-Sayre.
Alterazioni quantitative del mtDNA
Una riduzione della quantità del mtDNA è comunemente chiamata “deplezione”. Questa alterazione si associa comunemente a delle forme ad insorgenza infantile ad andamento progressivo. Gli organi più spesso coinvolti sono: muscolatura scheletrica e cardiaca, fegato e cervello. Le deplezioni del mtDNA sono causate da mutazioni in geni nucleari (vedi capitolo “mitocondriopatie dovute a mutazioni in geni nucleari”).
Mutazioni puntiformi del mtDNA.
Si tratta di quadri clinici associati a sostituzioni di singole basi o a micro-inserzioni/micro-delezioni, nella molecola del mtDNA. Queste mutazioni possono interessare sequenze codificanti RNA transfer (tRNA), RNA ribosomali (rRNA), o RNA messaggeri di proteine mitocondriali (mRNA). A differenza dei riarrangiamenti, che sono per lo più sporadici, quasi tutte le mutazioni puntiformi vengono trasmesse per via matrilineare. Spesso, ma non sempre, queste mutazioni sono eteroplasmiche. Sebbene, ad oggi, centinaia di mutazioni puntiformi siano state descritte in associazione con uno spettro estremamente eterogeneo di presentazioni cliniche, le mutazioni di gran lunga più frequenti sono solo quattro, e sono associate a sindromi cliniche piuttosto ben definite.
La neuropatia ottica ereditaria di Leber (Leber’s Hereditary Optic Neuropathy, LHON) (OMIM535000), ad esordio in età giovanile e netta prevalenza nel sesso maschile, è caratterizzata dalla perdita acuta o subacuta della visione centrale con esito in atrofia ottica. Il deficit visivo, che a parte rari casi è permanente, costituisce in genere l’unica manifestazione della malattia che però raramente comprende anche alterazioni del ritmo cardiaco (sindrome di pre-eccitazione ventricolare). La biopsia muscolare non evidenzia la presenza di fibre ragged-red, ed in effetti la biopsia non è in questo caso necessaria per la diagnosi. La malattia è stata associata a mutazioniù nelle posizioni nucleotidiche 3460, 11778 e 14484, che interessano rispettivamente le subunità ND1, ND4 e ND6 del complesso I della catena respiratoria. Altre mutazioni, sempre in geni del complesso I, sono state recentemente identificate. Molti aspetti della sindrome LHON rimangono da chiarire, quali l’estrema specificità tissutale delle lesioni cliniche, la prevalenza nel sesso maschile, e le conseguenze biochimiche di ciascuna mutazione.
La sindrome caratterizzata da neuropatia, atassia e retinite pigmentosa (Neurogenic muscle weakness, ataxia, retinitis pigmentosa, NARP) (OMIM551500) può comprendere, oltre ai suddetti segni, la epilessia e, talora, il decadimento mentale. La malattia ha in genere il suo esordio in età adulta. Anche in questo caso, la biopsia muscolare non mostra la presenza delle tipiche fibre ragged red. La malattia è stata associata alla mutazioneT8993G, nella sequenza codificante la subunità 6 dell’ATPasi mitocondriale (complesso V della catena respiratoria). Nella stessa posizione è inoltre descritta una transizione T->C presente in pazienti con fenotipo NARP meno grave. La stessa mutazione T8993G, con un grado di eteroplasmia nettamente più elevato rispetto al fenotipo NARP, è stata anche riscontrata in piccoli pazienti affetti da sindrome di Leigh (v. oltre) senza deficit biochimico di citocromo c ossidasi o piruvico deidrogenasi
La Encefalomiopatia Mitocondriale con Acidosi Lattica ed episodi simil-Stroke (Mitochondrial Encephalomyopathy, Lactic Acidosis and Strokelike episodes, MELAS) (OMIM540000) è definita dalla presenza delle seguenti manifestazioni: 1) episodi di tipo ictale (stroke-like) causati da lesioni cerebrali focali spesso localizzate nelle aree parieto-occipitali; 2) acidosi lattica o comunque livelli anormali di lattato nel sangue (e liquor); 3) fibre “ragged-red” nella biopsia muscolare. Altri segni di coinvolgimento del sistema nervoso centrale comprendono il deterioramento mentale, la cefalea ricorrente con vomito “cerebrale”, epilessia focale o generalizzata, e sordità neurosensoriale. La malattia è trasmessa per via materna e l’esordio è variabile, dalla primissima infanzia all’età giovanile-adulta.
La sindrome MELAS è tipicamente associata alla mutazione A3243G, nel gene codificante il tRNA per la Leucina (codone UUR). Sono state in seguito riportate altre mutazioni puntiformi associate a MELAS, anche se si tratta di casi più rari.
La mioclono epilessia con fibre ragged-red (Myoclonus Epilepsy with Ragged Red Fibers, MERRF) (OMIM545000) è caratterizzata dall’associazione di mioclono, epilessia, debolezza ed ipotrofia muscolare, incoordinazione motoria (atassia), e, talvolta, deterioramento mentale. L’entità delle manifestazioni cliniche può essere estremamente variabile nell’ambito della stessa famiglia. Tale variabilità si ritiene sia in relazione alla quantità di mtDNA mutato rispetto al normale (eteroplasmia) ed alla variabilità nella distribuzione tissutale della mutazione. La maggior parte delle famiglie affette è portatrice della transizione A8344G, nella sequenza del tRNA per la lisina.
Numerose altre mutazioni puntiformi del mtDNA sono state associate a diversi fenotipi clinici in singoli pazienti o in poche famiglie.
2. Mitocondriopatie dovute a mutazioni in geni nucleari
Oltre il 90% delle proteine mitocondriali sono espressione di geni nucleari. E’ perciò abbastanza sorprendente che, rispetto all’impressionante mole di osservazioni genetico-cliniche che riguardano alterazioni causate da mutazioni del mtDNA, malattie o sindromi causate da mutazioni in geni nucleari associati alla catena respiratoria siano finora relativamente scarse. D’altro canto, un numero crescente di malattie degenerative ereditarie, soprattutto in campo neurologico, si sono rivelate essere dovute a mutazioni in geni codificanti proteine mitocondriali, più o meno direttamente correlate alla OXPHOS.
I prodotti proteici codificati da questi geni possono essere raggruppati in tre categorie:
- 1. componenti strutturali della catena respiratoria
- 2. proteine che controllano l’OXPHOS o il metabolismo del mtDNA
- 3. proteine indirettamente correlate alla OXPHOS
|
Difetti di componenti strutturali della catena respiratoria* |
|
Difetto di complesso I ( Sindrome di Leigh, leucodistrofia, mioclono) |
|
Difetto di complesso II (Sindrome di Leigh, paraganglioma ereditario) |
|
Difetto di sintesi del Coenzima Q (Atassia, miopatia, crisi convulsive) |
|
Difetti di fattori che controllano l’OXPHOS o il metabolismo del mtDNA* |
|
Difetto di SURF1 (sindrome di Leigh) |
|
Difetto di SCO1 (encefalopatia infantile) |
|
Difetto di SCO2 (cardiomiopatia infantile) |
|
Difetto di COX10 (encefalopatia infantile) |
|
Difetto di COX15 (Cardiomiopatia Ipertrofica Fetale) |
|
Difetto di DGUOK (Sindrome da Deplezione di mtDNA, forma epatocerebrale) |
|
Difetto di TK2 (Sindrome da Deplezione di mtDNA, forma miopatica) |
|
Difetto di POLG1 (Oftalmoplegia Esterna Progressiva, Sindrome di Alpers) |
|
Difetto di BCS1 (encefalopatia infantile, tubulopatia, epatopatia) |
|
Difetto di ANT1 (oftalmoplegia esterna progressiva dominante) |
|
Difetto di Twinkle (oftalmoplegia esterna progressiva dominante) |
|
Difetto di Timidina Fosforilasi (encefalomiopatia neurogastrointestinale) |
|
Difetti di proteine indirettamente correlate ad OXPHOS |
|
Difetto di OPA1 (atrofia ottica dominante) |
|
Difetto di fratassina (atassia di Friedreich) |
|
Difetto di paraplegina (paraplegia spastica ereditaria) |
|
Difetto di Tim 8/9 (sordità, distonia, eredità legata X) |
* eredità mendeliana
Per brevità, accenniamo qui di seguito ad una sola malattia, la Sindrome di Leigh, perchè si tratta della malattia più nota e frequente del gruppo dei disordini “nucleari” dei mitocondri. I bambini affetti presentano, dopo un iniziale sviluppo normale nei primi mesi di vita, un progressivo ritardo dello sviluppo psicomotorio, accompagnato da incoordinazione dei movimenti oculari, vomito ricorrente, epilessia, anomalie del ritmo del respiro, e marcata acidosi lattica. In pratica sono soprattutto colpite le funzioni neurologiche che fanno capo ai sistemi che attraversano o sono localizzati nel tronco cerebrale e nel cervelletto.
Un’alterazione del metabolismo energetico mitocondriale è suggerita dal frequente aumento dei livelli di acido lattico nel siero e nel liquor.
In più della metà dei casi è possibile documentare un’alterazione genetica. In circa il 20% dei casi si tratta di mutazioni del mtDNA, per lo più nel gene dell’ATPasi 6 (mutazione NARP/MILS ma anche più rare mutazioni puntiformi in altre porzioni dello stesso gene). Più raramente si tratta di mutazioni del mtDNA che colpiscono geni codificanti le subunità della COX, o mutazioni in geni tRNA. In ca. il 30% dei casi il deficit biochimico principale è costituito da un difetto importante dell’attività del complesso IV (citocromo c ossidasi) e l’alterazione genetica è in questo caso dovuta, nella maggior parte dei casi, a mutazioni in un gene “assemblatore” del complesso IV, chiamato Surf-1. In altri casi il difetto biochimico è a carico del complesso I o del complesso II della Catena Respiratoria, e mutazioni delle subunità di questi complessi sono state in effetti identificate in alcuni pazienti. Infine, nel restante 10% dei casi di s. di Leigh è stato riscontrato un deficit di Piruvico Deidrogenasi, associato a mutazioni del gene legato al cromosoma X codificante la subunità E-1-alfa dell’enzima.
FORME NON SINDROMICHE.
Le forme non sindromiche sono caratterizzate da ipoacusia di grado lieve, moderato. pantonale, a volte con peggioramento sulle frequenze medie/centrali o acute, a trasmissione materna. Mutazioni a carico del DNA mitocondriale sono considerate responsabili di aumentata suscettibilità a farmaci (aminoglicosidi). rumore (noice-Induced hearing loss). età (aging-induced hearing loss). Il gene principalmente coinvolto nelle forme non sindromiche è MTRNR1 (12S rRNA) e la mutazione più frequente è A1555G (Tab. Van Camp 2010).
Mutazioni del DNA mitocondriale implicate nei casi di sordità non sindromiche
Il DNA mitocondriale è una parte di DNA di piccole dimensioni situata in ogni mitocondri. Esso codifica per 13 acidi ribonucleici messaggeri (mRNA), due RNA ribosomiali (rRNA) e 22 RNA di trasporto ed è trasmesso, unicamente delle madri, a tutti i loro figli. Normalmente la maggior parte degli individui normali ha un solo tipo di DNA mitocondriale (omoplasmia), ma possono essere presenti diversi tipi di DNA mitocondriale per tessuto (eteroplasmia), il che spiega per esempio la variabilità dei fenotipi tra fratelli, che in teoria dovrebbero essere colpiti nella loro totalità.
Sono state descritte diverse mutazioni nelle sordità isolate: la prima descritta e la più frequente, la mutazione A1555G dell’RNA ribosomiale 12S, [80] quindi le mutazioni C1494T dell’RNA ribosomiale 12S e T7511C, T7510C dell’RNA di trasferimento della serina.
A1555G è particolarmente frequente in Spagna nelle famiglie affette da sordità con trasmissione compatibile con una sordità mitocondriale, fino al 25%. [81] La prevalenza è scarsa in altri paesi. [82] La sordità dovuta alla mutazione A1555G può essere di ogni grado e apparire a qualunque età, spontaneamente o dopo assunzione di aminoglicosidi. [83] In effetti il bersaglio degli aminoglicosidi è l’RNA ribosomiale batterico e la forma dell’RNA ribosomiale 12S con mutazione A 1555 G diviene più vicina agli rRNA batterici che legano gli aminoglicosidi con un’affinità anormalmente elevata. Questo spiega la comparsa di sordità per dosi normali di aminoglicosidi in questi pazienti. Anche l’altra mutazione dell’RNA ribosomiale 12SC1494T può indurre anche una suscettibilità agli aminoglicosidi. [84, 85]
Forme non sindromiche
|
Gene |
Mutazioni |
Bibliografia
|
|
MTRNR1 |
1555A>G |
Prezant et al. 1993 |
|
MTRNR1 |
1494C->T |
Zhao et al. 2004 |
|
MTRNR1 |
961 |
Bacino et al , 1995 . Casano et al. 1999
|
|
MTTS1 |
7445A->G |
Reid et al . 1994
|
|
MTTS1 |
7472insC |
Tiranti et al. . 1995 |
|
MTTS1 |
75 10 T->C |
Hutchin et al. 2000 |
|
MTTS1 |
75 11 T->C |
Friedman et al.. 1999
|
Classificazione molecolare delle forme sindromiche e non, riconducibili a mutazioni mitocondriali A Martini ipoacusie infantili ed G.Paludetti
ED OMEGA 2011