RIASSUNTO
La sindrome branchio-oculo-facciale (BOFS) è caratterizzata da un
basso peso alla nascita e ritardo di crescita, schisi branchiali bilaterali che
possono essere emangiomatose, occasionali lesioni della cute dietro le orecchie
(lesioni simili a quelle prodotte da un’ustione), strabismo congenito, dotti
nasolacrimali ostruiti, radice nasale slargata con punta nasale appiattita,
labbro superiore sporgente con un filtro particolarmente ampio e prominente. Ad
oggi, sono stati descritti circa 50 casi. Altre malformazioni descritte
includono pseudoschisi del labbro superiore (che assomiglia ad una schisi
corretta chirurgicamente o una schisi fusa), padiglioni auricolari malformati,
la sordità di conduzione o neurosensoriale, le fossette preauricolari e
labiali, il palato ogivale, le anomalie dei denti, le anomalie oculari
(coloboma, microftalmia), e le cisti sottocutanee a livello del cuoio
capelluto. Negli adulti è presente incanutimento precoce. Un paziente
presentava, oltre alle caratteristiche tipiche della BOFS, agenesia parziale
del verme cerebellare, mentre due fratelli con questa sindrome avevano anche
cisti emangiomatose nelle orbite. L’esame urologico può rilevare anomali renali
(agenesia, cisti, idronefrosi). In due casi di BOFS sono stati descritti
polidattilia preassiale e frezza bianca dei capelli. L’intelligenza è normale e
la voce è ipernasale. La crescita persiste dopo la nascita, ma si mantiene tra
il terzo e il quinto percentile. I difetti branchiali cervicali retroauricolari
sono tra le caratteristiche più comuni della sindrome e in numerosi casi il
loro esame anatomopatologico evidenzia residui di timo. La BOFS è causata da
mutazioni che coinvolgono il gene TFAP2A (fattore di trascrizione alfa AP-2;
6p24). Anche se alcuni casi sono sporadici, la maggior parte dei casi descritti
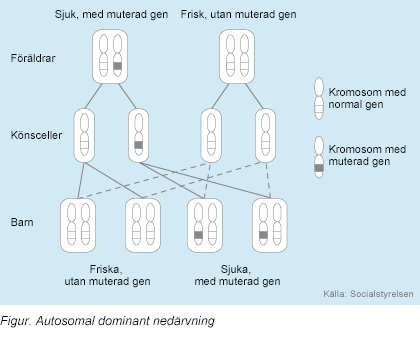
è familiare. La modalità di trasmissione è autosomica dominante. La diagnosi è
clinica e può essere confermata mediante analisi di DNA. La BOFS dovrebbe
essere differenziata dalla sindrome branchio-oto-renale (sindrome BOR; si veda
questo termine). La presa in carico è sintomatica e include un trattamento
combinato per la ipoacusia e per le lesioni cutanee. La prognosi è in funzione
della gravità delle manifestazioni associate.
La
sindrome branchio-oculo-facciale (BOFS) è caratterizzata da l’ubicazione anormale delle orecchie, aplastica lesioni
cutanee cervicali, padiglioni auricolari malformati, ipoacusia trasmissiva,
anomalie oculari, e labio-palatoschisi. La sindrome è stata descritta per la
prima nel 1982 da pediatri americani Woon Lee Kwang, Allen Root e Neil
Fenske. Un altro pediatra americano, Bryan Hall, ha descritto la
partnership con i propri dipendenti la stessa sindrome un anno più tardi.
Presenza incidenza
L’incidenza
non è nota. Le stime suggeriscono che la sindrome branchio-oculo-facciale
(BOFS) colpisce meno di una persona per milione. In Svezia noto per avere
un meno di 10 persone con la sindrome.
Causa
della malattia / infortunio
La
malattia è causata da un cambiamento (mutazione) nel gene TFAP2A sul
cromosoma 6 (6p24.3). TFAP2A è un modello per la produzione di
(codici) per il fattore di trascrizione AP-2 alfa. Un fattore di
trascrizione è una proteina che è essenziale per l’informazione nel DNA del
gene da trasferire (trascritto) all’RNA. DNA (acido desossiribonucleico) è
il nucleo della cellula e contiene le informazioni genetiche. RNA (acido
ribonucleico) è usato come messaggeri quando le proteine vengono
creati. TFAP2A richiesto per la trascrizione di geni che sono
importanti per lo sviluppo delle occhi, le orecchie e il viso durante il periodo
fetale precoce.
La
sindrome branchio-oculo-facciale (BOFS) è ereditata come carattere autosomico
dominante. Ciò significa che se un genitore ha la sindrome, cioè, avere un
gene normale ed un gene mutato, la probabilità di figli o figlie per farlo 50
percento. Quei bambini che non hanno avuto il gene mutato non possono
sindrome e per non oltre.
In
circa il 50-60 per cento ha la sindrome nasce come una mutazione
spontanea. La mutazione è poi di solito si è verificato in un gamete
genitoriale (ovuli o spermatozoi). La probabilità che essi saranno di
nuovo un bambino con la sindrome è quindi stimato a meno dell’1 per
cento. La mutazione di nuova provenienti da bambino, però, ereditaria e
può essere trasmessa alla generazione successiva.
La
sindrome branchio-oculo-facciale (BOFS) è caratterizzata da anomalie
craniofacciali, cervicali, oftalmologiche e orali. Possono includere dei
difetti del primo e secondo arco branchiale, può essere paragonata ad una
lesione cutanea aplastica o lineare angiomatosa, ed e stata descritta come
‘assenza congenita di cute’ orecchie; malformazioni degli occhi e delle
orecchie, labbro leporino e caratteristiche facciali particolari. La
gravità può variare da pochi e lievi anomalie di più approfonditi deformità e
disabilità.
Le
orecchie esterni possono essere deformati in modo diverso. Essi possono
essere più piccoli del normale (microtia), invertire ruotata e spesso sedersi
un po’più in basso sulla testa del normale. A volte, anche con
malformazioni del canale uditivo, orecchio medio e / o dell’orecchio interno,
che colpisce l’udito. Malformazioni delle orecchie esterne sono comuni
anche in altre sindromi e spesso, ma non sempre associato a una perdita
uditiva.
La
perdita dell’udito si verifica in circa il 70 per cento delle persone che hanno
la sindrome branchio-oculo-facciale (BOFS) . Può essere causato da perdita
uditiva causa di anomalie nel condotto uditivo e / o dell’orecchio medio
(ipoacusia), oa causa di cambiamenti dell’orecchio interno (sordità neurosensoriale),
ma può anche essere una forma combinata che include sia l’orecchio interno e
medio. Uditiva è dovuto a qualcosa che colpisce la capacità dell’orecchio
esterno o medio per trasmettere il suono al cervello e provoca una perdita
uditiva moderata. Quando il suono non viene fuori come dovrebbe sentire il
perdente ascoltando la nitidezza non è interessato. Malformazioni
dell’orecchio interno (coclea, coclea), significa che il worm ha meno
complicata rispetto alle fibre normali e nervose dell’orecchio interno non
funziona o mancante. Alcuni suoni sono distorti o scompaiono come tutto o
in parte.
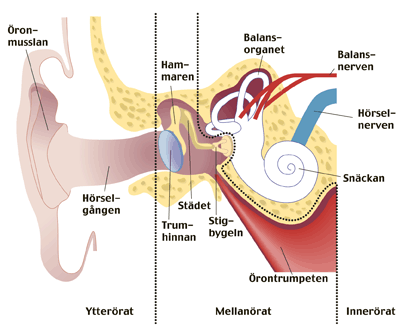
FIG. Il
suono è un’onda d’aria che provoca un movimento del timpano. Il movimento
propagata attraverso tre ossicini martello, incudine e staffa alla finestra ovale,
una membrana elastica che conduce l’organo dell’udito (coclea). Nella
coclea le cellule cigliate sono piegato quando l’onda sonora continua
attraverso un percorso di fluido, dopo di che la curva viene convertita in
impulsi elettrici. Il segnale viene poi inviato al nervo uditivo
attraverso i vari centri di commutazione nel cervello per il centro uditivo nei
lobi temporali.
Nelle
persone con sindrome branchio-oculo-facciale (BOFS) la perdita è spesso una ipoacusia
mista :trasmissiva e cocleare interno, ma i 2 differenti tipi possono essere
presenti anche separatamente. La perdita dell’udito è congenita e
inizialmente può essere da lieve a peggiorare lentamente, il che rende a volte
non viene rilevato fino a tardi. Si va da lieve a molto grave, e, talvolta,
può portare alla sordità.
L’organo
dell’i equilibrio che si trova adiacente alle coclea è deformato, influenzando
così l’equilibrio. Ciò potrebbe a sua volta portare a differenti abilità
motorie durante il primo anno di vita sono in ritardo (ad esempio, a rotolare,
sedersi e crawl), e che il bambino impara a camminare più tardi rispetto agli
altri bambini.
Le
malformazioni dell’orecchio a sindrome branchio-oculo-facciale (BOFS) sono
molto simili a quelle che si verificano in altre sindromi, come la sindrome di
22q11 delezione, sindrome Pendreds, sindrome branchiootorenale (BOR) e
brankiootosyndromet (BO)Per la sindrome branchio-oculo-facciale (BOFS) anomalie
sul collo e / o vicino alle orecchie (difetti della pelle
branchiali). Questi possono variare da spot con pelle sottile o aree dei capelli
coperti da fistole, aperture e cisti, che sono per lo più situati dietro le
orecchie e possono infettarsi.
La
maggior parte delle persone con la sindrome hanno anomalie negli occhi e nei
tessuti intorno agli occhi, come anormalmente piccoli occhi (microftalmia
unilaterale o piccole
fissurazioni palpebrali), o completa assenza di occhi (anoftalmia) e difetti di
chiusura (coloboma) dell’iride e della retina. Anomalie degli occhi
comporta un rischio per la perdita della vista. Si osservano anche Palpebre
cadenti (ptosi), restringimento dei dotti lacrimali, cataratta (cataratta) e lo
strabismo.
Labioschisi
con o senza palatoschisi, si verifica in quasi tutti gli individui con sindrome
branchio-oculo-facciale (BOFS) , ma palatoschisi isolatE sono
infrequenti. La faccia può anche avere altre caratteristiche estetiche
peculiari, come grandi occhi-set (ipertelorismo), aumentate la distanza tra gli
angoli degli occhi (telecanto), ampio punta del naso e bordi importanti sulle
ali nasali
Il Timo
(Thymus) potrebbe trovarsi al di fuori della sua posizione normale nella cavità
toracica (posizione ectopica). Circa in un terzo ci sono anche
malformazioni dei reni. Unghie e denti possono essere colpiti, e in alcuni
capelli diventano prematuramente grigi. A volte le persone hanno
polidattilia nelle mani e / o nei piedi ((Raveh E., Papsin BC, Forte V.
Branchio-oculo-facial syndrome. Int J Pediatr Otorhinolaryngol 2000 Jun
30;53(2):149-56).).
La
performance psicomotoria e solitamente normale, ma possono essere presenti
ritardo nello sviluppo, ipotonia e problemi di vista, di udito e di linguaggio.
(Lin-AE; Gorlin-RJ; Lurie-IW; Brunner-HG; vanderBurgt-I; Naumchik IV;
Rumyantseva-NV; StengelRutkowski-S; Rosenbaum-K; Meinecke-P; Muller-D. Further delineation of the
branchio-oculo-facial syndrome. AMERICAN-JOURNAL-OF-MEDICAL-GENETICS. MAR 13 1995; 56 (1) : 42-59)
.
Eziologia
La
sindrome branchio-oculo-facciale e una condizione che si trasmette con modalita
autosomica dominante.
Non e
ancora stato proposto un locus genico. Tuttavia, data la similitudine tra la
sindrome branchio-oculo-facciale e la sindrome branchio-oto-renale, ci si puo
aspettare che questo locus si trovi nel cromosoma 8q. (Raveh E., Papsin BC,
Forte V. Branchio-oculo-facial syndrome. Int J Pediatr Otorhinolaryngol 2000
Jun 30;53(2):149-56)
La
diagnosi viene fatta sulla base di segni esterni che hudavvikelser sul collo
alle orecchie, orecchie e occhi anomalie, perdita di udito, i tratti del viso
specifici. Fuori luogo timo e che ci sia un parente stretto con la
sindrome rafforza la sindrome di diagnosi brankiookulofacialt.
Malformazioni
dell’orecchio esterno devono sempre essere studiati perché a volte si
verificano in combinazione con, per esempio, il canale auricolare non è
completamente sviluppato; l’orecchio medio e / o coclea (coclea) sono deformate
o che il nervo uditivo manca.
Chiunque
sia sospettato di avere la sindrome è esaminato con la risonanza magnetica
(MRI) del orecchio e il nervo uditivo, per determinare la gravità della
malformazione dell’orecchio medio, orecchio interno e nervo uditivo è.
Il
grado di perdita dell’udito può essere determinata con diversi test dell’udito
(audiometria). Dal 2005, offerto a tutti i neonati in screening uditivo
Svezia, in connessione con la nascita. E ‘per mezzo di cosiddetti
otoemissioni acustiche (OAE), che verifica la funzione di cellule ciliate
esterne. Al tronco cerebrale, che è un metodo oggettivo per valutare il
grado di perdita dell’udito, il bambino testato per il ticchettio delle
cuffie. La risposta viene quindi registrato attraverso elettrodi nella
regione temporale. Tronco cerebrale misure se il segnale è passato dal
orecchio interno attraverso il nervo uditivo al centro uditivo del cervello. Audiometria
impedenza è utilizzato per esaminare le ossa dell’orecchio e la funzione
timpano dell’orecchio medio. Osservazione e gioco Condizionata audiometria dove
il bambino è, per esempio, ottenere blocchi in movimento quando si sente un
segnale o tono e discorso audiometria, che mette alla prova la capacità di
percepire toni o lingua parlata, sono altri metodi. Combinando i risultati
di queste misurazioni, la posizione e il grado di perdita dell’udito
determinati.
La
diagnosi viene confermata mediante analisi del DNA.
Quando
viene effettuata la diagnosi, è importante che alla famiglia sia offerta
consulenza genetica. La consulenza genetica include informazioni sulla
malattia e la sua eredità, valutazione basata albero genealogico analisi del
rischio di altri nella famiglia di ammalarsi, e informazioni sulla diagnosi e
il trattamento(Raveh E., Papsin BC, Forte V. Branchio-oculo-facial syndrome.
Int J Pediatr Otorhinolaryngol 2000 Jun 30;53(2):149-56). I vettori e
diagnostica fetale, così come la diagnosi genetica preimpianto (PGD) in
combinazione con la fecondazione in vitro, è possibile se la mutazione nella
famiglia è nota.
Non
esiste un trattamento che può curare la sindrome b branchio-oculo-facciale (BOFS) , senza
alcun sintomo trattati separatamente. Si può fare molto per sostenere e
compensare le limitazioni funzionali.
Se la
perdita dell’udito completamente o in parte dovuti malformazioni dell’orecchio
medio in alcuni casi è possibile operare. Apparecchi acustici può essere
utilizzato in entrambe le anomalie nell’orecchio interno e l’orecchio medio.
Alla
sordità da danni nell’orecchio interno, in alcuni casi impianto cocleare (CI)
utilizzati, ma sulla sindrome br branchio-oculo-facciale
(BOFS) deformità del verme volte essere così grande che non è possibile. L’impianto
cocleare t costituito da un processore vocale (un piccolo computer) che è posto
dietro l’orecchio e un impianto che viene chirurgicamente sotto la pelle, anche
dietro l’orecchio. L’impianto converte il suono in impulsi elettrici
codificati. I segnali vengono quindi trasmessi attraverso un elettrodo al
nervo uditivo, il cervello li interpreta come suono. I bambini che sono
nati sordi dovrebbero preferibilmente sottoporsi ad intervento chirurgico
intorno all’età di un anno. Una persona che una volta ascoltato, riesce a
ricordare i suoni e parole, se il vostro udito è deteriorata, e potrebbe
beneficiare di un impianto cocleare. E ‘importante che i bambini con
impianto cocleare offerti anche modi alternativi di comunicazione, come ad
esempio il segno.
Diverso
aspetto delle orecchie esterno può essere corretto, se necessario, la chirurgia
plastica. Se fistole o cisti al collo e sopra le orecchie causando
problemi, possono sottoporsi ad intervento chirurgico. Labbro leporino e
palato chirurgia precoce. È anche possibile operare zoccoli condotti uditivi
e condotti lacrimali.
Tratto
urinario dovrebbe essere esaminata con ecografia per vedere se ci sono
malformazioni renali. Continua follow-up medico può essere necessaria.
Malformazioni
oculari e anomalie degli occhi dovrebbero essere esaminati e controllati da un
oculista, che indaga la funzione visiva. Se ci sono coloboma o altre
anomalie oculari giudici oculista trattamento continuato. E ‘importante
per scoprire il caso di una anomalia unilaterale o bilaterale, perché è molto
importante per la visione. Si kolobomet situato in posizione centrale in
un occhio si scaricano molto ridotta acuità visiva, e ausili visivi
particolari. Coloboma VASCOLARE e la retina deve essere controllato
regolarmente per distacco di retina da rilevare in tempo e un deterioramento
della visione evitato.
I
bambini ei giovani che hanno una visione e l’udito ha bisogno abilitazione
della squadra con conoscenza approfondita di come la visione e dell’udito
perdita e la sordità influenzano l’interazione con l’ambiente e la
partecipazione alla vita sociale. Sforzi possono includere il trattamento,
la formazione, la consulenza e l’assistenza tecnica, sotto forma di aiuti di
vista / udito. Con l’aiuto di mezzi tecnici può aggiustamenti essere fatta
in casa e in età prescolare / scuola.
Supporto
e il trattamento è stato progettato in base alle esigenze che
esistono. Essi variano nel tempo e sono condotte in stretta collaborazione
con gli individui nel bambino / rete del giovanotto. La stretta
collaborazione si svolge anche con il Comune, che, sulla base del grado
d’invalidità in grado di offrire sostegno in varie forme per facilitare la vita
di tutti i giorni. Per incontrare altre famiglie con bambini nella stessa
situazione e imparare dalle reciproche esperienze sono utili.
Adulti
con sindrome branchio-oculo-facciale (BOFS) e problemi di vista e all’udito
bisogno di abilitazione in corso e tecnologie assistive. Casa e lavoro
potrebbero dover essere regolata mediante ausili visivi e acustici. Se si
dispone di un grado severo di visivo combinato e sentirlo contato come una
persona con sordocecità. Dove ci sono regionali sordociechi gestisce
l’abilitazione e la riabilitazione.
A
disabilità che hanno limitato capacità di lavoro può rivolgersi al Centro per
l’impiego per l’orientamento. Insurance Agency coordina gli sforzi
necessari per essere in grado di trovare o tornare al lavoro dopo una
disabilità influenzare lavoro.
–
HRF, HRF, Gävlegatan 16, Box 6605, 113 84 Stockholm, tel 08-457
55 00, textphone 08-475 55 01, fax 08-475 55 03, e-mail hrf@hrf.se, www.hrf.se .
Giovane udienza, Gävlegatan 16, Box 6605, 113 84 Stockholm, tel
08-457 55 00, textphone 08-475 55 01, fax 08-475 55 03,
e-mail kansli@uh.se,www.uh.se.
Associazione svedese dei Sordi, Rissneleden 138, 174 57
Sundbyberg, telefono 08-442 14 61, e-mail sdr@sdr.org, www.sdr.org.Svezia Sordi
Gioventù, Rissneleden 138, 174 57 Sundbyberg,
e-mailkansli@sduf.se, www.sduf.se.
Ipovedenti Associazione, Sandsborgsvägen 52 122 88 Enskede, tel
08-39 90 00, e-mail info@srf.nu, www.srf.nu.
Corsi, scambi di esperienze per il personale
–
Un’intensa attività di ricerca sulla sindrome di audizione
genetica andando in giro per il mondo. Molte delle sindromi di questo
gruppo sovrappongono e finora è stato difficile separare, ma è ora possibile
per un gran numero per diagnostica genetica. Collaborazione di ricerca si
svolge tra i dipartimenti genetiche degli ospedali universitari.
Per ogni messaggio di diagnostica nel database Consiglio sulle
malattie rare è un breve riassunto in forma illustrativo. Gli opuscoli
possono essere scaricati e stampati (vedere “di più per noi” nella
colonna di destra).
Abbo O, Bieth E, Ballouhey Q, Vaysse F, Just W, Galinier P.
Branchi-oculo-facial syndrome: a case report to highlight recent genetic
considerations. J Plast Reconstr Aesthet Surg 2012; 65: 1573-1575.
Al-Dosari MS, Almazyad M, Al-Ebdi L, Mohamed JY, Al-Dahmash S,
Al-Dhibi H et al. Ocular manifestations of branchio-oculo-facial syndrome:
report of a novel mutation and review of the literature. Mol Vis 2010; 16:
813-818.
Carter MT, Blaser S, Papsin B, Meschino W Reardon W, Klatt R et
al. Middle and inner ear malformations in mutation-proven branchio-oculo-facial
(BOF) syndrome: case series and review of the literature. Am J Med Genet A
2012; 8: 1977-1981.
Galliani E, Burglen L, Kadlub N, Just W Sznajer Y, de Villemeur
TB et al. Craniofacial phenotype in the branchio-oculo-facial syndrome: four case
reports. Cleft Palate Craniofac J 2012; 49: 357-364.
Hall BD, de Lorimier A, Foster LH. Brief clinical report: a new
syndrome of hemangiomatous branchial clefts, lip pseudoclefts, and unusual
facial appearance. Am J Med Genet 1983; 14: 135-138.
Lee WK, Root AW, Fenske N. Bilateral branchial cleft sinuses
associated with intrauterine and postnatal growth retardation, premature aging,
and unusual facial appearance: a new syndrome with dominant transmission. Am J
Med Genet 1982; 11: 345-352.
Lin AE, Gorlin RJ, Lurie IW, Brunner HG van der Burgt I,
Naumchik IV, Rumyantseva NV, Stengel-Rutkowski S et al. Further delineation of
the branchio-oculo-facial syndrome. Am J Med Genet 1995; 56: 42-59.
Lin AE, Yuzuriha S, McLean S, Mulliken JB. Lesser forms of cleft
lip associated with the branchio-oculo-facial syndrome. J Craniofac Surg 2009;
20: 608-611.
Milunsky JM, Maher TM, Zhao G, Wang Z, Mulliken JB, Chitayat D
et al. Genotype-phenotype analysis of the branchio-oculo-facial syndrome. Am J
med Genet A 2011; 155: 22-32.
Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT
et al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum
Genet 2008; 82: 1171-1177.
Thomeer HG, Crins TT, Kamsteeg EJ, Buijsman W, Cruysberg JR,
Knoers NV et al. Clinical presentation and the presence of hearing impairment
in branchio-oculo-facial syndrome: a new mutation in the TFAP2A gene. Ann Otol
Rhinol Laryngol 2010; 119: 806-814.
OMIM (Online Mendelian Inheritance in Man)
www.ncbi.nlm.nih.gov/omim
Sökord: branchiooculofacial syndrome, BOFS
GeneReviews (University of Washington)
www.ncbi.nlm.nih.gov/books/NBK1116
Sökord: branchiooculofacial syndrome, BOF syndrome
GeneReviews ® [Internet].
Termine di ricerca
![]()
·
GeneReviews
Ricerca Avanzata
·
Aiuto
Sindrome
Branchiooculofacial
Sinonimo:
Sindrome BOF
Angela
e Lin, MD, FAAP, FACMG e Jeff M Milunsky, MD, FACMG.
Distacco iniziale: 31
maggio 2011.
Sommario
Caratteristiche cliniche.
La sindrome Branchiooculofaciale
(BOFs) è caratterizzata da: branchiale (cervicale [90%] o [60%] infrastrutture
o sovra-auricolare) difetti della pelle che spaziano dalla pelle sottile appena
percettibile o patch di capelli per le lesioni eritematose
“emangiomatose” al grande pianto erosioni; anomalie oculari che
possono includere microftalmia, anoftalmia, coloboma e stenosi/ atresia del condotto
nasolacrimale ; le anomalie facciali possono includere l’ipertelorismo
oculare o telecanto, punta larga del naso, rime palpebrali upslanted, labbro
leporino o pilastri philtral importanti che danno l’aspetto di un labbro
leporino riparato (precedentemente chiamato “labbro pseudocleft”) con
o senza palatoschisi, labbro superiore pozzi e debolezza facciale inferiore
(asimmetrica faccia piangere o parziale 7 ° debolezza
nervo cranico). Padiglioni auricolari e udito perdita malformati e di
primo piano da orecchio interno e / o anomalie rocca petrosa sono comuni. L’intelletto
è di solito normale.
Diagnosi / testing.
La diagnosi si basa sul
quadro clinico. TFAP2A è l’unico gene in cui mutazioni
sono attualmente noti per causare BOFs.
Gestione.
Trattamento delle
manifestazioni: In generale, i bambini con BOFs dovrebbero essere gestiti da
un team multi specialistico compresi, ad esempio, gli specialisti
cranio-facciali, terapisti chirurghi plastici, otorinolaringoiatri, e del
linguaggio. Piccole, lineari o superficiali difetti della buccia
branchiali possono guarire spontaneamente; tuttavia, alcuni richiedono un
intervento chirurgico. Anoftalmia o microftalmia grave può richiedere un
con formero (una struttura, solitamente di plastica, inserite nella cavità
oculare per favorire la sua crescita); stenosi del dotto naso-lacrimale o
l’atresia spesso richiede un intervento chirurgico. Si raccomanda di
labbro leporino essere riparato da un pediatra chirurgo plastico
esperto. Forme minori di labioschisi (“pseudocleft”) possono
avere bisogno di correzione chirurgica.
Sorveglianza: Monitor per
variazioni relative ai principali risultati nel tempo.
Consulenza genetica.
BOFs è ereditata
come autosomica dominante maniera. De
novo mutazioni sono osservate nel 50% -60% dei colpiti individui. Ogni
figlio di un individuo con BOFs ha una probabilità del 50% di ereditare
la mutazione. La diagnosi prenatale per gravidanze a rischio è
possibile se la mutazione responsabile
della malattia è stato identificato un membro di famiglia colpita.
Diagnosi
Sfondo. La sindrome
branchiooculofacial (BOFs) è una sindrome cranio-facciale distintivo primo
caratterizzato da Lee et al [1982], Hall et al [1983], e Fujimoto et al [1987]. Lin et al [1995] ha pubblicato una
grande revisione del risultati clinici. L’associato gene (TFAP2A) è
stato scoperto nel 2008 utilizzando array di ibridazione
genomica comparativa [Milunsky et al 2008], seguita da una grande genotipo – fenotipo analisi[Milunsky et al 2011].
Diagnosi clinica
La sindrome
branchiooculofacial (BOFs) viene diagnosticata clinicamente. Non ci sono
linee guida diagnostiche formali sviluppate da pannelli di consenso,
utilizzando algoritmi di una gerarchia di dati clinici, o standard di test
basati sull’evidenza.
I criteri diagnostici sono
stati utilizzati in modo informale in base ai difetti caratteristici (B, O, F)
e sono stati formalmente proposto nel 2011 che incorpora l’importanza delle
anomalie del timo e diagnosticato in modo indipendente parenti di primo
grado [Milunsky et al
2011 Tabella
I)].
Nota: Dei tre
caratteristiche originali che compongono il BOF mnemonico, la “B”
(difetto pelle cutanea) è il più caratteristico quando è bilaterale e cervicali
anteriori in posizione.
Criteri diagnostici:
- Tutti e tre delle principali
caratteristiche sono presenti:- Branchiale (cutanea) difetto di
pelle - Anomalia oculare
- Anomalie facciali (caratteristico
aspetto del viso) O
- Branchiale (cutanea) difetto di
- Due dei tre principali
caratteristiche più uno dei seguenti sono presenti:- Colpiti parente di
primo grado, diagnosticato
in modo indipendente - Timo ectopica (cutanea)
- Colpiti parente di
Nota: i risultati puntati
elencati di seguito, quelli in grassetto sono presenti nella
maggior parte colpiti individui; risultati
in corsivo non sono presenti negli individui più colpiti, ma
si distinguono per BOFs.
Branchiali (cutanee)
difetti
- Cervicali (90%) o infrastrutture
o sovra-auricolare (60%) i difetti della pelle - Variare da pelle sottile appena
percettibile o patch di capelli per eritematosa “emangiomatose”
lesioni alle grandi erosioni piangenti - Differire dai tratti punteggiano
seno della branchiootorenal (BOR) Sindrome - Difetti più lievi possono essere riconosciuti,
guariscono spontaneamente
Anomalie oculari
- Microftalmia, anoftalmia
- Coloboma
- Strabismo
- Ptosi
- Stenosi del dotto naso-lacrimale
/ atresia - Cataratta
Anomalie facciali
- Aspetto caratteristico con dolicocefalia,
ipertelorismo o telecanto, larga punta del naso, rime palpebrali
upslanted (Figura1) - Labioschisi (o pilastri
philtral importanti tecnicamente noto come labbro leporino forma minore(precedentemente
chiamato “labbro pseudocleft”), con o
senza palatoschisi (99%), ma non isolatopalatoschisi - Pozzi labbro superiore
- Del nervo facciale inferiore e /
o ipoplasia muscolare (asimmetrica faccia piangere, parziale 7 ° debolezza
nervo cranico) - Dell’orecchio interno e delle
ossa petrosa anomalie (come nella sindrome CHARGE e la sindrome dibranchiootorenal, non ci può
essere la displasia cocleare, Mondini displasia, e ampliato acquedotto
vestibolare.) - Padiglioni auricolari malformati
e prominente - La perdita dell’udito (70%)
(conduttiva, neurosensoriale, misto)

Foto di un bambino di cinque anni con la sindrome BOF. I
dettagli delle scoperte molecolari sono riportate nelle Milunsky et al [2008]
(3 pazienti, all’età di 2 anni). Ha un difetto cutaneo cervicale destro
lati (“B”), che è stato riparato; dotto naso-lacrimale
bilaterale (more
…)
Sistema immunitario
- Anomalie timo (ectopica, derma)
Sistema renale
- Anomalie strutturali (35%)
(displastici, assente, multicistica, etc.) - Reflusso vescico-ureterale
Ectodermica (capelli,
denti, unghie)
- Incanutimento prematuro (poliosi) (35%)
- Denti ipoplasiche
- Chiodi displasiche
- Cisti (spesso sul cuoio
capelluto, meno comunemente nella regione testa e del collo)
Sviluppo psicomotorio
- Handicap visivi e uditivi
(frequente) - Performance psicomotoria (di
solito normale) - L’autismo disturbi
dello spettro, disabilità
intellettiva (raro)
Crescita
- Restrizione della crescita: non
comuni
Rari (<5 pazienti)
- Iridi eterocromia
- Cardiopatia congenita (difetto
del setto atriale, tetralogia di Fallot) - Polidattilia (bilaterali, di
solito post-assiale) - Medulloblastoma (descritto una
volta [Milunsky et al 2008])
Molecolare Test genetici
Gene. TFAP2A è l’unico gene in cui mutazione è noto per provocare
la sindrome branchiooculofacial.
Sperimentazione clinica
- Analisi di sequenza. Più del 95% degli
individui che soddisfano i criteri diagnostici clinici per BOFs haTFAP2A sequenza
varianti rilevabili mediante sequenza di analisi (cioè, piccole intragenica
delezioni / inserzioni e missense, nonsense, e sito di splice
mutazioni) [Milunsky et al 2011]. - Cancellazione / duplicazione analisi. All’in-
grosso gene delezioni di TFAP2A sono
stati descritti[Milunsky et al 2008, Gestri et al 2009] e probabilmente sono
presenti in meno del 5% delle persone con BOFs. Delezioni più grandi
di cromosoma 6p24-p25 compresi TFAP2A sono
rari [Davies et al 1999,Misceo et al 2008]. Non sono stati
riportati parziale gene o exonic delezioni o duplicazioni.
Tabella 1.
Sintesi
di ricerca di genetica molecolare Utilizzato nella sindrome Branchiooculofacial
|
Gene 1 |
Metodo di prova |
Mutazioni |
L’identificazione |
|
TFAP2A |
Le analisi della |
Sequenza varianti 4 |
> 95% |
|
Cancellazione /duplicazione analisi 5 |
All’in- grosso genedelezioni |
<5% |
1.
Vedi Tabella Geni A. e
database per cromosoma locus e proteine.
2.
Vedere Genetica Molecolare per informazioni su
varianti alleliche.
3.
La
capacità del metodo utilizzato per rilevare una mutazione che è presente nella
indicata gene
4.
Esempi
di mutazioni identificate da analisi di sequenza possono includere
piccole intragenica delezioni / inserzioni e missense, nonsense, e sito di
splice mutazioni; tipicamente, exonic o all’in- grosso gene delezioni /
duplicazioni non vengono rilevati. Per problemi da considerare
nell’interpretazione dei risultati delle analisi di sequenza, fare clic qui.
5.
Test
che identifica delezioni / duplicazioni non facilmente rilevabili
mediante analisi della sequenza di codifica e
regioni fiancheggianti intronic di genomico del DNA; inclusi nella
varietà di metodi che possono essere utilizzati sono: PCRquantitativa, a lungo raggio PCR,
multiplex ligation-dipendente probe amplificazione
(MLPA), e cromosomica microarray(CMA) a cui questo gene / cromosoma segmento.
Strategia dei saggi
Per confermare / stabilire
la diagnosi in un probando
1.
L’esame
clinico per le funzioni di diagnostica
2.
Per
quei che soddisfano i criteri diagnostici clinici, l’analisi della sequenza dei sette esoni
codificanti eintrone / esone confini TFAP2A
3.
Se
una mutazione non viene rilevato
sulla analisi della sequenza, cancellazione / analisi
di duplicazione
La diagnosi prenatale e
diagnosi genetica preimpianto (PGD) per gravidanze a rischio richiedono previa identificazione
della mutazione patogenetica in famiglia.
Geneticamente Correlate
(alleliche) Disturbi
Nessun altro fenotipi sono
noti per essere associati con la mutazione di TFAP2A.
Caratteristiche cliniche
Descrizione clinica
Femmine e maschi
sono colpiti allo stesso modo.
I neonati con crepacci hanno
le sfide che ci si attende per l’alimentazione, ed eventualmente la
respirazione.
I neonati con fessure hanno
bisogno di supporto per una corretta alimentazione, e fare meglio quando nella cura
di un team di palatoschisi stabilito. Alcuni dei più piccoli difetti della
pelle cominciano a “curare” per seconda intenzione. Stenosi del
dotto naso-lacrimale o l’atresia portare a occhi piangenti, e richiedono
l’attenzione di un oculista pediatrico.
Durante l’infanzia, gli
individui con BOFs stanno affrontando con cosmetici, visive, uditive, e le
sfide di discorso. Possono avere lo strabismo; alcuni hanno
significativa compromissione visiva. I più gravementecolpiti i bambini hanno
chirurgici, medici, cosmetici e di apprendimento ha bisogno di simile a molti
altri bambini con disturbi cranio-facciali. Tuttavia, i bambini lievemente
affetti possono apparire ad avere bisogno di poco sostegno.
Gli adolescenti possono avere
problemi con l’ansia e la stima sociale. Questi sembrano essere più che
l’esperienza di vivere con un labbro leporino e altre anomalie cranio-facciali,
ma il numero di affetti individui è piccolo.
La maggior parte hanno
l’intelletto normale.
Adulti con BOFs tipiche
sono state di solito diagnosticata durante l’infanzia. Gli individui con
caratteristiche molto lievi possono non essere diagnosticati fino a che non
danno vita a un classico colpito bambino.L’osservazione
che gli adulti possono essere asintomatica o minimamente influenzato illustra
l’espressione variabile delle mutazioni in questo gene.
Fertilità non sembra
essere influenzata.
Genotipo-fenotipo
Correlazioni
Genotipo / fenotipo correlazioni per mutazioni
all’interno TFAP2A non sono ben stabiliti.
A genotipo – fenotipo analisi di 41
individui in 30 famiglie si è basata su criteri diagnostici che sono state
soddisfatte in 87% [Milunsky et al 2011]. Inter-
significativo e variabilità
intrafamiliare sono stati osservati con le stesse mutazioni [Milunsky et al 2011]. Missense,
frameshift, e splicing mutazioni insieme
con riarrangiamenti complessi più [Tekin et al 2009, Milunsky
et al 2011] in tutto il gene risultato in
fenotipi simili.
L’assenza di clefting
evidente si nota nei pochi individui che hanno all’in- grosso gene delezioni [Milunsky et al2008, Gestri et al 2009] e più grandi
delezioni cromosomiche che includono TFAP2A [Davies et
al 1999,Misceo et al 2008]. Tutti gli
individui con delezione sembrano avere un
philtrum anormalmente importante che potrebbe essere sullo spettro di
microforma labioschisi [Lin et al 2009]. In caso
contrario, la inter- e marcatavariabilità
intrafamiliare sembra simile a quella osservata con mutazioni intragenica.
Fino ad oggi le informazioni
sono insufficienti per fare delle generalizzazioni sulla presenza di tratti
autistici e grave ritardo mentale in BOFs da solo due casi sono stati notati.
- SP2 paziente riportata da Reiber et al [2010], che aveva la mutazione c.806T> C, aveva sia
grave ritardo mentale e autismo. - Paziente 3 riportata da Gestri et al [2009], che ha avuto un
all’in- grosso gene delezione, aveva
caratteristiche autistiche.
Penetranza
BOFs ha mostrato quasi
completa penetranza. Un attento esame di individui identificati in
una famiglia con BOFs con un TFAP2A mutazione è necessario per
rivelare i risultati sottili tra cui incanutimento precoce (individui possono
aver tinto i capelli), capelli debole sul collo, o eterocromia delle iridi.
Anticipazione
Dati insufficienti sono stati
segnalati per valutare se l’anticipazione può verificarsi in
BOFs. Questo è particolarmente difficile a causa della variabilità clinica
marcata in questo disturbo. Cinque delle sei famiglie con BOFs senza mosaicismo identificato aveva
apparentemente più “gravi” scoperte nelle generazioni successive[Milunsky et al 2011]. Ulteriore
studio è necessario.
Nomenclatura
Il termine “sindrome
branchiooculofacial” è stato suggerito da Fujimoto et al [1987]. Nessun altro
nome è stato usato per questa sindrome distintivo.
Prevalenza
BOFs è rara, con meno di
100 casi ben descritti, con analisi di genetica molecolare. La prevalenza
non è nota.
Diagnosi differenziale
Sovrapposizione fenotipica
con il branchiootorenal (BOR) sindrome è stato
suggerito, ma BOFs ha più distintive anomalie cranio-facciali, e le due cose
non devono essere confusi.
Anche se ci può sembrare
superficiale sovrapposizione alcuni dei risultati, non ci dovrebbe essere
alcuna confusione nel differenziare BOFs da sindrome CHARGE in quanto
quest’ultimo non ha difetti della buccia, e BOFs non avere atresia delle
coane. Il padiglione auricolare e dell’orecchio interno anomalie anormali
differiscono.
Gestione
Le valutazioni dopo la
diagnosi iniziale
Per stabilire l’entità
della malattia e le esigenze di un individuo con diagnosi di sindrome branchiooculofacial
(BOFs), si consigliano i seguenti studi:
- CT Imaging dell’osso temporale di
anticipare correzione ottimale audizione [Raveh et al 2000, Tekin et al2009, Stoetzel et al 2009] - Ecocardiogramma se vi è un
mormorio cardiaco o sintomi - L’ecografia renale
I seguenti consultazioni sono
consigliati:
- L’esame dei difetti cutanei da un
chirurgo plastico pediatrico per delineare l’estensione della lesione (s),
per determinare se vi è un seno e, soprattutto, per determinare se un
residuo timica potrebbe essere presente - Valutazione formale di labio /
palatoschisi e altre possibili anomalie facciali da un team labbro /
palatoschisi, che spesso include un genetista medico, pediatrico chirurgo
plastico, otorinolaringoiatra, logopedista, specialista dentale e
ortodonzia, e oculista - Visita oculistica completa da un
oculista pediatrico per valutare limitazioni visive, lo strabismo, e
ostruzione del dotto naso-lacrimale - Rinvio di quelli con anoftalmia e
/ o grave microphthalmia ai servizi di supporto per i non vedenti - Rinvio ad un nefrologo se sono identificate
anomalie renali - Rinvio ad un audiologo
- Per quelli con una fenditura, la
valutazione da un logopedista - Valutazione Development
soprattutto per i bambini con problemi visivi e / o acustici - Monitoraggio per la depressione,
l’attenzione disregolazione
Nota: i ritardi motore non
fanno parte di BOFs, e, quindi, la terapia fisica e occupazionale non è
prevedibile.
Trattamento delle
manifestazioni
Milunsky et al [2011] ha fornito linee
guida di gestione. Vedere Linee guida.
In generale, i bambini con
BOFs e molteplici anomalie dovrebbero essere seguiti in un ambiente in cui la
cura multispecialty può essere fornita da un team tra cui, per esempio,
specialisti cranio-facciali, chirurghi plastici, otorinolaringoiatri e
logopedisti [adattato da Milunsky
et al 2011 Tabella III ].
Idealmente, le valutazioni
multispecialty e chirurgia devono essere eseguite all’interno di una clinica
cranio-facciale. Il trattamento chirurgico deve essere fatto solo da un
chirurgo plastico pediatrico esperto nel trattamento labbro
leporino. Forme minori di labioschisi (precedentemente conosciuto come
“pseudocleft”) possono avere bisogno di correzione chirurgica [Lin et al 2009]. Oltre al
appiattimento punta nasale o asimmetria che possono essere associati con il
labbro leporino, una caratteristica completa, punta del naso piatta può avere
bisogno di una procedura correttiva. Inoltre colpiti gli individui
possono avere bisogno di ricostruzione di malformati padiglioni auricolari
sporgenti. Se diagnosticato nella prima infanzia, fusione auricolare può
essere indicato.
Quando i difetti della
pelle branchiali o sovra-auricolari sono piccole, lineari, o superficiale,
possono guarire spontaneamente. I difetti della buccia più grandi
dovrebbero essere trattati come una “ferita” umida da un chirurgo
plastico, ma in genere non hanno bisogno di un intervento chirurgico. Non
dovrebbero essere cauterizzato. La maggior parte dei difetti della buccia
più grandi richiedono l’escissione chirurgica. È importante sottolineare
che un tratto del seno deve essere sezionato da un pediatra chirurgo plastico
esperto. Esplorazione per un residuo timica può essere necessario, che
deve essere inviato per l’esame istopatologico.
Ostruzione da stenosi del
dotto nasolacrimale o atresia deve essere sollevato e monitorato per
restenosi.Microftalmia grave o anoftalmia possono essere gestite con
l’inserimento di un conformero nella cavità oculare per favorire la sua
crescita.
La perdita dell’udito è
trattata di routine (vedi sordità ereditaria e
dell’udito Panoramica perdita).
I denti devono essere
monitorati per dimensioni e numero, la carie, e malocclusione.
Sfide sensoriali,
psicologici, e di sviluppo devono essere trattati con terapie di
supporto. Al momento, i dati sono insufficienti per fornire
raccomandazioni che più gravemente colpiti individui richiedono
un supporto più psicologica.
Sorveglianza
Monitor per variazioni
relative ai principali risultati nel tempo.
Monitorare i bambini più
grandi che entrano nell’adolescenza segni di scarsa autostima e di altre
questioni psicologiche.
Valutazione del Parenti
a rischio
Vedere consulenza genetica per le questioni
legate alla sperimentazione di a rischio parenti per consulenza genetica scopi.
Terapie Sotto inchiesta
Cerca ClinicalTrials.gov per l’accesso alle informazioni su studi clinici per una
vasta gamma di malattie e condizioni. Nota: Non ci possono essere gli
studi clinici per questo disturbo.
Consulenza genetica
La consulenza genetica è
il processo di fornitura di individui e famiglie con informazioni sulla natura,
l’ereditarietà, e le implicazioni di malattie genetiche per aiutarli a prendere
decisioni personali e mediche informate. I seguenti sezione tratta la
valutazione del rischio genetico e l’uso della storia familiare e test genetici
per chiarire lo status genetico per i familiari. Questa sezione non è
destinata a affrontare tutte le questioni personali, culturali, etiche o che
gli individui possono affrontare o per sostituire la consultazione con una
genetica professionali. -ED.
Modalità di eredità
La sindrome
Branchiooculofacial (BOFs) è ereditata come autosomica dominante maniera.
Rischio per i familiari
I genitori di un probando
- Una percentuale significativa (~
40% -50%) di individui con diagnosi di BOFs hanno un affetto genitore[Milunsky et al 2011]. - Un probando con BOFs può avere la
malattia come risultato di de novo mutazione. La
proporzione di casi provocati da de novo mutazione si
avvicina al 50% -60% [Milunsky et al 2011]. - Si raccomanda che i genitori di
un probando hanno test di genetica molecolare per la variante patogena
specifica alle famiglie per facilitare accurato rischio di
ricorrenza di
consulenza. La valutazione dei genitori può stabilire che si è colpiti, ma è
sfuggito precedente diagnosi a causa della mancata da personale sanitario
a riconoscere la sindrome e / o di una presentazione fenotipica più
lieve. Pertanto,
un apparentemente negativa storia di famiglia non può essere confermata
fino a quando il test molecolare è stato completato.
Nota: (1) Anche se ~ 40%
-50% delle persone con diagnosi di BOFs hanno un affetto genitore, la storia della famiglia può sembrare essere
negativo a causa del mancato riconoscimento del disturbo di membri della
famiglia o morte precoce del genitore prima della manifestazione di sintomi (ad
esempio, incanutimento precoce). (2) Se il genitore è l’individuo in cui
la mutazione prima verificato s /
egli può avere somatica mosaicismo per la mutazione e
può essere leggermente / minimamente interessato.
Fratelli e sorelle di
un probando
- Il rischio per i fratelli
del probando dipende dallo stato
genetico dei genitori del probando. - Se un genitore del probando è interessata e / o ha un TFAP2A variante
patogena, il rischio per i fratelli e sorelle di ereditare la variante
patogena è del 50%. - Quando i genitori sono clinicamente inalterati (dopo attento esame), il
rischio per i fratelli e sorelle di unprobando sembra essere basso. - I fratelli e sorelle di un probando clinicamente non affetti genitori sono ancora ad
aumentato rischio per BOFs a causa della possibilità di espressività
variabile e
/ o mosaicismo
germinale in
un genitore.
Prole di un probando. Ogni figlio di un
individuo con BOFs ha una probabilità del 50% di ereditare la variante
patogena.
Altri membri della
famiglia di un probando. Il rischio per gli altri membri della famiglia dipende dallo
stato dei genitori del probando. Se un parente viene colpita, i suoi
familiari possono essere a rischio.
Genetica Related Issues
Counseling
Per determinare se un
genitore, figlio o altro membro della famiglia potrebbero essere colpiti, un attento
esame fisico è il “test”. Più importante
Considerazioni in famiglie
con un apparente de novo mutazione. Quando nessuno dei
genitori di unprobando con una autosomica dominante condizione ha
evidenza clinica del disturbo, è probabile che i probandi ha una de
novo mutazione. Tuttavia, la variabile espressività di BOFs warrants
test molecolare dei genitori per i più accurato rischio di ricorrenza di
consulenza. Possibili spiegazioni non medici, tra cui la paternità alternativo o di maternità (ad
esempio, con la riproduzione assistita) o l’adozione riservate potrebbero anche
essere esplorate.
Pianificazione famigliare
- Il momento ottimale per la
determinazione del rischio genetico e discussione della disponibilità di
test prenatale è prima della gravidanza. - È opportuno offrire una consulenza
genetica (compresa
la discussione dei rischi potenziali per la prole e le opzioni di
riproduzione) a giovani adulti che sono interessati.
Banking DNA è la conservazione
di DNA (tipicamente estratto da globuli bianchi) per un possibile uso
futuro.Poiché è probabile che la metodologia di prova e la nostra comprensione
dei geni, varianti alleliche, e le malattie migliorerà in futuro, occorre
tenere in considerazione per bancaria DNA di affetti individui.
Test prenatale
Test di genetica
molecolare. La
diagnosi prenatale per gravidanze a rischio è possibile mediante l’analisi diDNA estratto da cellule
fetali ottenuti mediante l’amniocentesi solito eseguita a circa 15 a 18
settimane di gestazione o prelievo dei villi coriali (CVS) a circa dieci a 12
settimane di gestazione. La variante allelica patogena di un colpiti membro della
famiglia deve essere identificato nella famiglia prima del test prenatale può
essere eseguita.
Esame ecografico. Una revisione
sistematica delle caratteristiche prenatali di BOFs Non è stato
condotto. E ‘possibile che la diagnosi prenatale di labio / palatoschisi
in un feto a maggior rischio di BOFs permetterebbe la sindrome di essere
diagnosticata.
Nota: l’età gestazionale è
espresso in settimane mestruali calcolate sia a partire dal primo giorno
dell’ultimo periodo mestruale normale o da misure ad ultrasuoni.
Diagnosi genetica
preimpianto (PGD) può essere un’opzione per le famiglie nelle quali è stata
identificata la variante patogena.
Risorse
Personale GeneReviews ha
selezionato le seguenti organizzazioni malattia-specifici e / o di supporto
ombrello e / o registri per il beneficio di soggetti con questo disturbo e le
loro famiglie. GeneReviews non è responsabile per le informazioni fornite
da altre organizzazioni. Per informazioni sui criteri di selezione,
clicca qui.
- AboutFace Internazionale
123 Edward Street
Suite 1003
Toronto Ontario M5G 1E2
Canada
Telefono: 800-665-3223 (numero
verde); 416-597-2229
Fax: 416-597-8494
Email: info@aboutfaceinternational.org
www.aboutfaceinternational.org
- AmeriFace: fessura / avvocati
cranio-facciali
PO Box 751112
Las Vegas NV 89136
Telefono: 888-486-1209 (numero
verde 24 ore); 702-769-9264
Fax: 702-341-5351
Email: info@ameriface.org
Genetica molecolare
Le informazioni contenute
in tabelle Genetica Molecolare e OMIM può differire da quello in altre parti
del GeneReview: tavoli può contenere informazioni più recenti. – ED.
Tabella A.
Sindrome
Branchiooculofacial: Geni e database
|
Gene |
Cromosoma Locus |
Proteina |
Locus specifico |
HGMD |
I
dati sono compilati dai seguenti riferimenti standard: gene
da HGNC; cromosoma locus, nome luogo, regione critica,
complementazione gruppo da OMIM; proteine
da UniProt. Per una descrizione di database (Locus
specifico, HGMD) a cui vengono forniti i link, clicca qui.
Tabella B.
OMIM
iscrizioni per la sindrome Branchiooculofacial (Vedi tutti in OMIM)
|
FATTORE DI TRASCRIZIONE |
|
|
SINDROME |
Genetica molecolare
Patogenesi
TFAP2A è un membro di
acido-sensibile retinoico della famiglia AP-2 di trascrizione fattori che
regolano geneespressione durante l’embriogenesi degli
occhi, orecchie, viso, parete del corpo, degli arti, e del tubo neurale[Schorle et al 1996, Zhang et al 1996, Ahituv
et al 2004, Nelson & Williams 2004].
Gene struttura. TFAP2A contiene sette esoni
codificanti (riferimento della sequenza NM_003220.2). Per un
riepilogo dettagliato delle gene informazioni e
proteine, vedi tabella A, Gene.
Varianti alleliche
patogeni. Le
mutazioni all’interno TFAP2A o la cancellazione di tutto il gene risultato in
branchiooculofacial (BOF) sindrome. Milunsky
et al [2008] hanno descritto un famigliare delezione intero
gene e quattro de novo mutazioni missense in casi simplex
(cioè, una singola occorrenza in famiglia) che ha provocato
BOFs. Ulteriori mutazioni e un’altra eliminazione familiare sono stati ora
descritti [Gestri et al 2009,Stoetzel et al 2009, Tekin et al 2009, Reiber
et al 2010].
Anche se le mutazioni
avvengono in tutto il gene, una regione
hotspot negli esoni 4 e 5 che ospita mutazioni missense in circa il 90% dei
probandi / famiglie con BOFs è stato identificato [Milunsky et al 2011]. Ricorrenti
mutazioni sono ormai riconosciuti (Tabella 2).
Mosaicismo è stato
rilevato in una famiglia [Milunsky et al 2011].
Lo spettro molecolare in
30 famiglie con 41 colpite individui con BOFs
incluso mutazioni eterozigoti missense (28/30; 93%), una mutazione frameshift, e uno
all’in- grosso gene delezione [Milunsky et
al 2011.] Tekin et al [2009] ha segnalato un
complesso TFAP2A allele (soppressione di 18
e l’inserimento di 6 nucleotidi) tra
gli amminoacidi 276 e 281 in un individuo con BOFs.
Tabella 2.
Recurrent TFAP2A patogeno
Varianti Rivelando una mutazionale Hotspot
|
Exon |
DNA Nucleotide Change |
Proteine |
Sequenze di |
|
4 |
c.709C> G |
p.Arg237Gly |
|
|
4 |
c.710G> C |
p.Arg237Pro |
|
|
4 |
c.724G> A |
p.Glu242Lys |
|
|
4 |
c.752G> A |
p.Gly251Glu |
|
|
4 |
c.760A> G |
p.Arg254Gly |
|
|
4 |
c.760A> T |
p.Arg254Trp |
|
|
4 |
c.761G> C |
p.Arg254Pro |
|
|
4 |
c.763A> G |
p.Arg255Gly |
|
|
5 |
c.767C> T |
p.Ala256Val |
Tabella
adattata da tabella III in Milunsky
et al [2011].
Nota
sulla classificazione variante: Varianti elencate nella tabella sono stati
forniti dagli autori. GeneReviews personale non ha verificato
in modo indipendente la classificazione delle varianti.
Nota
sulla nomenclatura: GeneReviews segue le convenzioni di
denominazione standard del Human Genome Variation Society(www .hgvs.org). Vedere di riferimento rapido per una spiegazione
di nomenclatura.
Normale gene prodotto. Proteina TFAP2A
comprende 437 aminoacidi. Ha una centrale di base di DNAregione di legame, un
terminale carbossi motivo elica-portata-elica che media dimerizzazione, e un
amino-terminale che contiene un dominio di transattivazione [Eckert et al 2005]. Gli aminoacidi
nella regione di base del dominio di legame al DNA (esoni 4 e 5) mostrano
elevata conservazione evolutiva da Homo sapiensattraverso Ciona
intestinalis (trasparente ascidia) [Milunsky et al 2008].
Oltre al suo ruolo nella
regolazione genica espressione durante
l’embriogenesi, TFAP2A si occupa anche di tumorigenesi
con espressione proteica livelli che
interessano la trasformazione cellulare, la crescita del tumore, metastasi e la
sopravvivenza [Jean et al 1998, Heimberger
et al 2005, Orso et al 2007]. Numerose
interazioni gene probabilmente alla base della variabilità del fenotipo risultante da
difetti molecolari che coinvolgono TFAP2A. TFAP2A è noto per essere
espresso in cellule della cresta neurale premigratory e migratori [Hilger-Eversheim et al 2000, Li & Cornell, 2007] ed è necessario
per la morfogenesi precoce della lente [Gestri et al2009].
Anormale gene prodotto. Negli esseri umani,
le anomalie descritte in BOFs sembra essere correlato a mutazioni o delezioni
di TFAP2A portano alla regolamentazione disfunzionale
soprattutto durante l’embriogenesi.
La perdita o l’alterazione
della funzione di TFAP2A ortologhi proteine in zebrafish o topi
provocano clefting facciale, anomalie degli arti, e difetti dell’occhio,
orecchio, parete del corpo, del tubo neurale, e deflusso cuore tratto [Schorle et al 1996, Zhang et al 1996, Nottoli
et al 1998, Sud-Mays ed altri 1999, Brewer
et al 2002,Holzschuh et al 2003, Cavaliere et al 2003, Ahituv
et al 2004, Brewer
et al 2004, Nelson & Williams 2004,Feng et al 2008]. Gestri et al [2009] ha studiato il ruolo
di TFAP2A mutazione in zebrafish
morfogenesi occhio che ha rivelato un’associazione con una moltitudine di
patologie oculari. Inoltre, le mutazioni compromessa lagene funzione di
sensibilizzare in tal modo l’occhio in via di sviluppo ad una mutazione
deleteria di altri geni, tra cui BMP4 e tcf711a [Gestri et al 2009]. Damberg
[2005] ha rilevato che la famiglia AP-2 può essere coinvolto nella regolazione
della sistemi monoaminergici nel cervello adulto, causando disordini
neuropsichiatrici. Brewer et al [2004] ha osservato che sopravvivono mutanti
Tcfap2a topi hanno anomalie cranio-facciali, anomalo sviluppo dell’orecchio
medio, e difetti di pigmentazione.
Riferimenti
References
Literature Cited
- Ahituv
N, Erven A, Fuchs H, Guy K, Ashery-Padan R, Williams T, de Angelis MH,
Avraham KB, Steel KP. An ENU-induced mutation in AP-2alpha leads to middle
ear and ocular defects in Doarad mice. Mamm Genome. 2004;15:424–32. [PubMed] - Brewer
S, Jiang X, Donaldson S, Williams T, Sucov HM. Requirement for AP-2alpha
in cardiac outflow tract morphogenesis. Mech
Dev. 2002;110:139–49. [PubMed] - Brewer
S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of
AP-2alpha causes multiple neural crest-related defects. Dev
Biol. 2004;267:135–52. [PubMed] - Damberg
M. Transcription factor AP-2 and monoaminergic functions in the central
nervous system. J
Neural Transm. 2005;112:1281–96. [PubMed] - Davies
AF, Mirza G, Flinter F, Ragoussis J. An interstitial deletion of 6p24-p25
proximal to the FKHL7 locus and including AP-2alpha that affects anterior
eye chamber development. J
Med Genet.1999;36:708–10. [PMC free article] [PubMed] - Eckert
D, Buhl S, Weber S, Jäger R, Schorle H. The AP-2 family of transcription
factors. Genome
Biol.2005;6:246. [PMC free article] [PubMed] - Feng
W, Huang J, Zhang J, Williams T. Identification and analysis of a
conserved Tcfap2a intronic enhancer element required for expression in
facial and limb bud mesenchyme. Mol
Cell Biol.2008;28:315–25. [PMC free article] [PubMed] - Fujimoto
A, Lipson M, Lacro RV, Shinno NW, Boelter WD, Jones KL, Wilson MG. New
autosomal dominant branchio-oculo-facial syndrome. Am J Med Genet. 1987;27:943–51. [PubMed] - Gestri
G, Osborne RJ, Wyatt AW, Gerrelli D, Gribble S, Stewart H, Fryer A, Bunyan
DJ, Prescott K, Collin JR, Fitzgerald T, Robinson D, Carter NP, Wilson SW,
Ragge NK. Reduced TFAP2A function causes variable optic fissure closure
and retinal defects and sensitizes eye development to mutations in other
morphogenetic regulators. Hum
Genet. 2009;126:791–803. [PMC free article] [PubMed] - Hall
BD, deLorimier A, Foster LH. Brief clinical report: a new syndrome of
hemangiomatous branchial clefts, lip pseudoclefts, and unusual facial
appearance. Am
J Med Genet. 1983;14:135–8. [PubMed] - Heimberger
AB, McGary EC, Suki D, Ruiz M, Wang H, Fuller GN, Bar-Eli M. Loss of the
AP-2alpha transcription factor is associated with the grade of human
gliomas. Clin
Cancer Res. 2005;11:267–72.[PubMed] - Hilger-Eversheim
K, Moser M, Schorle H, Buettner R. Regulatory roles of AP-2 transcription
factors in vertebrate development, apoptosis and cell-cycle control. Gene. 2000;260:1–12. [PubMed] - Holzschuh
J, Barrallo-Gimeno A, Ettl AK, Durr K, Knapik EW, Driever W. Noradrenergic
neurons in the zebrafish hindbrain are induced by retinoic acid and
require tfap2a for expression of the neurotransmitter phenotype. Development. 2003;130:5741–54. [PubMed] - Jean
D, Gershenwald JE, Huang S, Luca M, Hudson MJ, Tainsky MA, Bar-Eli M. Loss
of AP-2 results in up-regulation of MCAM/MUC18 and an increase in tumor
growth and metastasis of human melanoma cells. J Biol
Chem. 1998;273:16501–8. [PubMed] - Knight
RD, Nair S, Nelson SS, Afshar A, Javidan Y, Geisler R, Rauch GJ, Schilling
TF. lockjaw encodes a zebrafish tfap2a required for early neural crest
development. Development. 2003;130:5755–68. [PubMed] - Lee
WK, Root AW, Fenske N. Bilateral branchial cleft sinuses associated with
intrauterine and postnatal growth retardation, premature aging, and
unusual facial appearance: a new syndrome with dominant
transmission. Am
J Med Genet. 1982;11:345–52. [PubMed] - Li
W, Cornell RA. Redundant activities of Tfap2a and Tfap2c are required for
neural crest induction and development of other non-neural ectoderm
derivatives in zebrafish embryos. Dev Biol. 2007;304:338–54. [PMC free article] [PubMed] - Lin AE,
Gorlin RJ, Lurie IW, Brunner HG, van der Burgt I, Naumchik IV, Rumyantseva
NV, Stengel-Rutkowski S, Rosenbaum K, Meinecke P. et al. Further delineation of
the branchio-oculo-facial syndrome. Am J Med Genet. 1995;56:42–59. [PubMed] - Lin
AE, Yuzuriha S, McLean S, Mulliken JB. Lesser forms of cleft lip
associated with the branchio-oculo-facial syndrome. J Craniofac
Surg. 2009;20 Suppl 1:608–11. [PubMed] - Milunsky
JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, Burch MN, Clemens
M, Mulliken JB, Smith R, Lin AE. TFAP2A mutations result in
branchio-oculo-facial syndrome. Am J Hum Genet.2008;82:1171–7. [PMC free article] [PubMed] - Milunsky
JM, Maher TM, Zhao G, Wang Z, Mulliken JB, Chitayat D, Clemens M, Stalker
HJ, Bauer M, Burch M, Chénier S, Cunningham ML, Drack AV, Janssens S,
Karlea A, Klatt R, Kini U, Klein O, Lachmeijer AM, Megarbane A, Mendelsohn
NJ, Meschino WS, Mortier GR, Parkash S, Ray CR, Roberts A, Roberts A,
Reardon W, Schnur RE, Smith R, Splitt M, Tezcan K, Whiteford ML, Wong DA,
Zori R, Lin AE. Genotype-phenotype
analysis of the branchio-oculo-facial syndrome. Am J Med Genet
A. 2011;155A:22–32. [PubMed] - Misceo
D, Bjørgo K, Ormerod E, Ringen Ø, Rocchi M, van der Hagen CB, Frengen E. A
de novo 6p interstitial deletion and a complex translocation involving
chromosomes 2, 6, and 14 in a mildly developmentally delayed
patient. Am J
Med Genet A. 2008;146A:3230–3. [PubMed] - Nelson
DK, Williams T. Frontonasal process-specific disruption of AP-2alpha
results in postnatal midfacial hypoplasia, vascular anomalies, and nasal
cavity defects. Dev
Biol. 2004;267:72–92. [PubMed] - Nottoli
T, Hagopian-Donaldson S, Zhang J, Perkins A, Williams T. AP-2-null cells
disrupt morphogenesis of the eye, face, and limbs in chimeric
mice. Proc Natl Acad Sci U S A. 1998;95:13714–9. [PMC free
article]
[PubMed] - Orso
F, Fassetta M, Penna E, Solero A, De Filippo K, Sismondi P, De Bortoli M,
Taverna D. The AP-2alpha transcription factor regulates tumor cell
migration and apoptosis. Adv
Exp Med Biol.2007;604:87–95. [PubMed] - Raveh E,
Papsin BC, Forte V. Branchio-oculo-facial syndrome. Int J Pediatr
Otorhinolaryngol.2000;53:149–56. [PubMed] - Reiber
J, Sznajer Y, Posteguillo EG, Müller D, Lyonnet S, Baumann C, Just W.
Additional clinical and molecular analyses of TFAP2A in patients with the
branchio-oculo-facial syndrome. Am
J Med Genet A.2010;152A:994–9. [PubMed] - Schorle
H, Meier P, Buchert M, Jaenisch R, Mitchell PJ. Transcription factor AP-2
essential for cranial closure and craniofacial development. Nature. 1996;381:235–8. [PubMed] - Stoetzel
C, Riehm S, Bennouna Greene V, Pelletier V, Vigneron J, Leheup B, Marion
V, Hellé S, Danse JM, Thibault C, Moulinier L, Veillon F, Dollfus H.
Confirmation of TFAP2A gene involvement in branchio-oculo-facial syndrome
(BOFS) and report of temporal bone anomalies. Am J Med Genet
A.2009;149A:2141–6. [PubMed] - Tekin
M, Sirmaci A, Yüksel-Konuk B, Fitoz S, Sennaroğlu L. A complex TFAP2A
allele is associated with branchio-oculo-facial syndrome and inner ear
malformation in a deaf child. Am
J Med Genet A.2009;149A:427–30. [PubMed] - West-Mays
JA, Zhang J, Nottoli T, Hagopian-Donaldson S, Libby D, Strissel KJ,
Williams T. AP-2alpha transcription factor is required for early
morphogenesis of the lens vesicle. Dev Biol.1999;206:46–62. [PubMed] - Zhang
J, Hagopian-Donaldson S, Serbedzija G, Elsemore J, Plehn-Dujowich D,
McMahon AP, Flavell RA, Williams T. Neural tube, skeletal and body wall
defects in mice lacking transcription factor
AP-2.Nature. 1996;381:238–41. [PubMed]
Chapter Notes
Author Notes
As of January,
2011, there is no disease advocacy organization (“support group”) for BOFS.
Through Dr. Lin, several parents of children with BOFS have reached out to the
families of newly diagnosed individuals.
Acknowledgments
We thank the
many families and international colleagues who have supported our research.
Revision History
- 31 May 2011 (me) Review posted
live - 11
January 2011 (al) Original submission
Copyright ©
1993-2015, University of Washington, Seattle. All rights reserved.
For more
information, see the GeneReviews Copyright Notice and Usage
Disclaimer.
For questions
regarding permissions: ude.wu@tssamda.
Bookshelf ID: NBK55063PMID: 21634087

Ann
Dermatol. 2009 Aug; 21 (3): 288-290. Inglese.
Pubblicato online il 31 agosto 2009. http://dx.doi.org/10.5021/ad.2009.21.3.288
Copyright ©
2009 coreano dermatologica Associazione e la Società coreana per Dermatology
Investigative
Un
caso di sindrome Branchio-oculo-facciale
Min Young Park, MD e Lei Chan Kim, MD![]()
Dipartimento di Dermatologia, Ajou University
School of Medicine, Suwon, Corea.
![]() Ristampa
Ristampa
richiesta: Si Chan Kim, MD, Dipartimento di Dermatologia, Ajou University
School of Medicine, 5, Woncheon-dong, Yeongtong-gu, Suwon 443-721, Corea.Tel:
82-31-219-5190, Fax: 82-31-219-5189, E-mail:maychan@ajou.ac.kr
Ricevuto 12
Dicembre 2008; Accettato 8 Gennaio 2009.
Questo è un
articolo Open Access distribuito sotto i termini della licenza Creative Commons
Attribuzione-Non commerciale (http://creativecommons.org/licenses/by-nc/3.0) che
consente libero uso non commerciale, la distribuzione e la riproduzione in
qualsiasi medie, purché l’opera originale sia correttamente citata.
A cinque
anni di età, il paziente ha visitato la nostra clinica dermatologia per le
lesioni del collo persistenti. Le lesioni cutanee erano più guarito
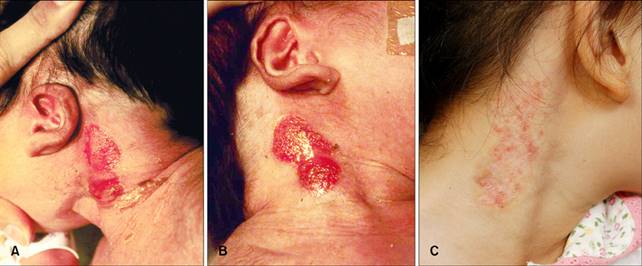
rispetto al passato, ma zone poco epitelizzata rimaste (Fig. 1C). Campioni bioptici pelle hanno mostrato caratteristiche
di guarigione pelle con epidermide appiattite, fibrosi nel derma e totale
assenza di strutture annessiali (Fig. 2B). I genitori hanno riferito che la riparazione chirurgica
del labbro leporino e sonda l’inserimento di ostruzione dotto naso-lacrimale
sono state eseguite a 10 mesi di età. Inoltre, durante il primo anno di
vita, una cisti è stato trovato sul cuoio capelluto posteriori, che è stato
rimosso. Le lesioni del collo sono stati trattati in modo conservativo con
una semplice medicazione. Si consiglia di rivalutazione degli occhi,
orecchie e reni, e la riparazione chirurgica per le lesioni
cutanee. Tuttavia, i genitori hanno rifiutato un’ulteriore valutazione e
trattamento invasivo. Il trattamento ha incluso l’applicazione della
soluzione di fattore di crescita epidermico attualità e idrocolloidi
spogliatoio materiale.
DISCUSSIONE
BOFs è una
malattia genetica rara caratterizzata dalla presenza di cranio-facciale
distintivo, cervicale, auricolare, e le anomalie oftalmologiche. Questi
possono includere i difetti del seno branchiali con pelle aplastica, basso
delle orecchie con rotazione posteriore, labioschisi con o senza palatoschisi,
una pseudocleft del labbro superiore, ostruzione del dotto naso lacrimale e
microftalmia 2.Sebbene i criteri diagnostici non sono stati chiaramente
definiti, le caratteristiche di cui sopra quando presenti insieme solito
consentono per una diagnosi clinica. Inoltre, anomalie renali, tra cui
l’agenesia, aplasia, ipoplasia o displasia dei reni non sono
infrequenti. In alcuni casi, la perdita dell’udito conduttivo, ectopica
dermica timo, cisti del cuoio capelluto, e anomalie ectodermiche come piccoli
denti e le unghie displastiche sono anche noti 3. La maggior parte di
queste caratteristiche sono stati trovati nel nostro caso. BOFs viene
ereditata come una malattia autosomica dominante a penetranza incompleta e
espressione variabile4. I cromosomi appaiono normali, e non locus genico è
stato identificato. Tuttavia, recentemente, Milunsky et al. 5riferito che
BOFs è causata da mutazioni che coinvolgono il gene TFAP2A, che è uno della
famiglia AP-2 di fattori di trascrizione. Questo gene è stato dimostrato
che regolano lo sviluppo di protuberanze facciali, gemme degli arti, chiusura
cranica e la vescicola lente. Uno studio genetico non è stato condotto nel
nostro caso perché i suoi genitori non erano d’accordo di avere lo studio
fatto. Tuttavia, finora la diagnosi di BOFs è basata sulle manifestazioni
cliniche e non sulla identificazione di un gene mutato 6. Pertanto,
si considera che una diagnosi di BOFs era ragionevole in questo caso.
La diagnosi
differenziale del BOFs comprende sindrome branchiooto-renale
(BORS). Entrambi possono essere associati con stenosi nasolacrimale
condotto, perdita di udito, e anomalie renali. Tuttavia, le
caratteristiche cliniche quali la labiopalatoschisi, pseudocleft del labbro, e
la pelle aplastica non si trovano in BORS. Inoltre, i geni responsabili
per i due sindromi sembrano essere diverso 7.
La gestione
di BOFs deve essere individualizzato sulla base delle anomalie
associate. In alcuni casi, le lesioni cutanee aplastiche di BOFs,
spontaneamente regrediscono lasciando una lesione cicatrice simile
a 8; in altri casi sono necessari escissione chirurgica e riparazione 9. Quando
presente, il timo dermica ectopica può esistere sul lato con la lesione
aplastica; la presenza di un timo normale radiografia toracica deve essere
confermata prima di rimuovere la lesione 10.
References
1.
Lee WK, Root AW, Fenske N. Bilateral branchial cleft sinuses
associated with intrauterine and postnatal growth retardation, premature aging,
and unusual facial appearance: a new syndrome with dominant transmission. Am J Med
Genet 1982;11:345–352.
![]()
![]()
2.
Hall BD, deLorimier A, Foster LH. Brief clinical report: a new
syndrome of hemangiomatous branchial clefts, lip pseudoclefts, and unusual
facial appearance. Am J Med Genet 1983;14:135–138.
![]()
![]()
3.
Lin AE, Gorlin RJ, Lurie IW, Brunner HG, van der Burgt I, Naumchik
IV, et al. Further delineation of the branchio-oculo-facial syndrome. Am J Med
Genet 1995;56:42–59.
![]()
![]()
4.
Fujimoto A, Lipson M, Lacro RV, Shinno NW, Boelter WD, Jones KL,
et al. New autosomal dominant branchio-oculo-facial syndrome. Am J Med
Genet 1987;27:943–951.
![]()
![]()
5.
Milunsky JM, Maher TA, Zhao G, Roberts AE, Stalker HJ, Zori RT, et
al. TFAP2A mutations result in branchio-oculo-facial syndrome. Am J Hum Genet
2008;82:1171–1177.
![]()
![]()
6.
Raveh E, Papsin BC, Forte V. Branchio-oculo-facial syndrome. Int J
Pediatr Otorhinolaryngol 2000;53:149–156.
![]()
![]()
7.
Trummer T, Muller D, Schulze A, Vogel W, Just W.
Branchio-oculo-facial syndrome and branchio-otic/branchio-oto-renal syndromes
are distinct entities. J Med Genet 2002;39:71–73.
![]()
![]()
8.
El Darouti MA, Marzouk SA, Azzam OA, Nada HR, Sobhi RM, El
Nabarawi I. Branchio-oculo-facial syndrome with bilateral linear scars of the
neck. Int J Dermatol 2005;44:674–676.
![]()
![]()
9.
Hiroshi F, Satoru S, Eisuke U, Kunihiro K, Yuhei Y. Bilateral
dermal thymus of neck in branchio-oculo-facial syndrome. J Plast
Reconstr Aesthet Surg 2006;59:1385–1387.
![]()
10. Civi
I, Kurtay M, Civi S. Bilateral thymus found in association with unilateral
cleft lip and palate. Plast Reconstr Surg 1989;83:143–147.
![]()
![]()
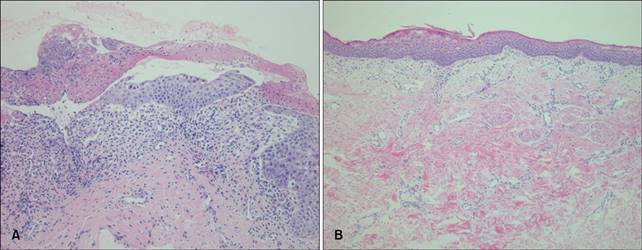
una biopsia
cutanea dalla lesione collo del neonato mostrava un’ulcera con derma infiltrati
infiammato e completa assenza di strutture appendageal. (B) La pelle di
guarigione dimostrato epidermide appiattite con fibrosi cutanea. Strutture
annessiali non sono stati rispettati (A, B: H & E, x 200).
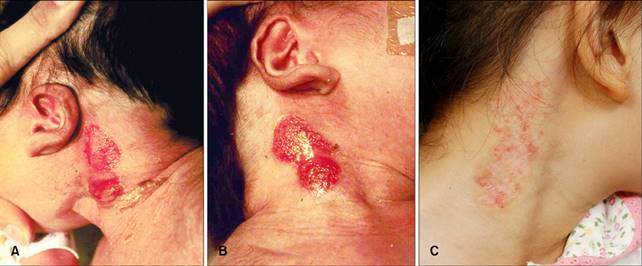
(A, B) Il neonato si è presentato con patch
erosivi su entrambi i lati del collo. Sono evidenti pure delle orecchie basse,
con rotazione posteriore. (C) Cinque anni dopo, la lesione aveva ancora la
pelle poco guarito con riepitelizzazione incompleta.