MELAS (Mitochondrial Encephalomyopathy, Lactic, Acidosis, and Stroke-like episodes)
Riassunto
La sindrome MELAS (encefalomiopatia mitocondriale con
acidosi lattica e episodi simili a ictus) è una patologia progressiva
caratterizzata da disturbi neurologici acuti paragonabili a ischemie cerebrali,
associati a iperlactatemia e miopatia mitocondriale. La prevalenza è
sconosciuta. Durante l’infanzia o la prima adolescenza, i pazienti sono
soggetti di solito a crisi acute, forse causate da un’infezione o da uno sforzo
fisico. Queste crisi si associano a cefalee, vomito e a volte ad episodi
pseudoischemici, emiparesi e emianopsia. Si manifestano spesso in pazienti con
sintomi cronici come deficit motorio, sordità, diabete, bassa statura,
cardiomiopatia, ritardo dello sviluppo, difficoltà di apprendimento, di memoria
e di attenzione. La malattia è dovuta alle mutazioni del DNA mitocondriale.
Sono state identificate 10 mutazioni differenti ma l’80% dei casi è dovuto alla
mutazione 3243A>G nel gene del tRNA della leucina (tRNA Leu). Questa
mutazione viene definita spesso mutazione MELAS, sebbene sia associata a segni
clinici diversi; la sua prevalenza in Europa è stimata in 1 su 6250 e si
associa alla sindrome in oltre il 7,5% dei pazienti. La diagnosi si basa sulle
manifestazioni cliniche e sulla risonanza magnetica cerebrale. La risonanza
magnetica può rivelare la presenza di numerose lesioni profonde nella materia
grigia e bianca del cervello, mentre la TAC identifica atrofia cerebrale e
calcificazioni dei gangli basali. La risonanza e la TAC mostrano che le lesioni
non sono confinate nelle aree vascolari e di conseguenza gli episodi acuti non
si possono considerare tipici ictus. La concentrazione anomala di lattato è
frequente nel sangue e pressoché costante nel liquido cerebrospinale. La
biopsia muscolare è anomala nell’85% dei pazienti, mostra una proliferazione
mitocondriale atipica (fibre rosse lacerate) e fibre muscolari con deficit di
citocromo-ossidasi. L’analisi delle attività enzimatiche della catena
respiratoria muscolare può rivelare un deficit del complesso I o un deficit
combinato dei complessi I e IV. L’identificazione della mutazione causale deve
tenere conto della sua costante eteroplasmia, cioè la sua coesistenza con una
popolazione residuale di DNA mitocondriale normale. La proporzione delle
mutazioni varia notevolmente a seconda dei tessuti, ma spesso può essere molto
elevata (oltre il 90% della popolazione del DNA mitocondriale) e può essere
ricercata anche nel sangue. La terapia è complicata dall’eteroplasmia. La
mutazione è trasmessa per eredità materna. Un maschio affetto non è in grado di
trasmettere la malattia. Sebbene una proporzione elevata della mutazione nel
sangue della madre aumenti il rischio di nascita di un bambino gravemente
affetto dalla malattia, esistono molti esempi di segregazione estrema
madre-figlio, che rendono difficile la consulenza genetica. La presenza di
percentuali eterogenee della mutazione nei diversi tessuti impedisce, in
teoria, la diagnosi prenatale. Sono state effettuate poche sperimentazioni
cliniche. Una sperimentazione recente ha scoperto che il dicloroacetato ha un
effetto deleterio nel medio termine. L’evoluzione spontanea, fatta di crisi
seguite da recuperi e ricadute, rende difficile valutare il miglioramento in
alcuni pazienti sottoposti a trattamenti (che utilizzano il coenzima Q10 e
l’analogo idebenone, la creatina monoidrato e l’arginina) o il peggioramento
dovuto ad alcuni trattamenti come l’acido valproico (farmaco a effetto
antiepilettico responsabile di episodi simili a ictus). La prognosi è grave.
Gli episodi potrebbero provocare il decesso del paziente e la loro ricorrenza
nel lungo periodo può causare un deterioramento mentale, perdita della vista e
dell’udito e grave miopatia, che potenzialmente può contribuire alla perdita
dell’autonomia
Che cosa è la MELAS
L’encefalopatia
mitocondriale, acidosi lattica con episodi tipo ictus –
abbreviato MELAS
(miopatia, encefalopatia, acidosi lattica, e
episodi di simil-ictus) – è una
della famiglia di citopatie mitocondriali , che comprendono anche MERRF , e neuropatia ottica ereditaria di Leber , è una
malattia neurodegenerativa progressiva caratterizzata da episodi neurologici
acuti simili a tratti associati iperlattatemia e miopatia mitocondriale.
È stata per la prima volta
caratterizzata con questo nome nel 1984 da Pavlakis SG ET AL.[1] Una caratteristica di queste malattie è
che sono causate da difetti del DNA nel genoma mitocondriale che viene ereditata esclusivamente dal genitore femminile[Hirano
M, Pavlakis SG,1994].[2]. [ 2 ] Tuttavia, è importante sapere che alcuni delle proteine
essenziali per la normale funzione mitocondriale sono prodotte
dal genoma nucleare, e vengono successivamente trasportati ai mitocondri per
uso.Come tale, le mutazioni in queste proteine possono provocare
malattie mitocondriali, ma può essere ereditato da entrambi i genitori maschi e
femmine nel modo tipico. La malattia può manifestarsi in entrambi i sessi.
INCIDENZA
La prevalenza esatta della malattia è
sconosciuta. I pazienti di solito presentano durante l’infanzia o la prima
età adulta con crisi acute, che possono essere attivati da
infezione o l’esercizio fisico. Queste crisi socio cefalea, vomito e
talvolta segni pseudo-ictus, come confusione, emiparesi e
emianopsia. Spesso si verificano in pazienti con sintomi cronici come la
debolezza muscolare, sordità, diabete, bassa statura, cardiomiopatia, ritardo
dello sviluppo, difficoltà di apprendimento, perdita di memoria o disturbi
dell’attenzione.
SEGNI E SINTOMI
MELAS è una condizione che colpisce molti dei sistemi del corpo,
in particolare il cervello e il sistema nervoso (encefalopatia) e muscoli
(miopatia). Nella maggior parte dei casi, i segni ed i sintomi di questa
malattia compaiono durante l’infanzia, dopo un periodo di normale
sviluppo. [ 3 ] I primi sintomi
possono includere debolezza muscolare e dolore, mal di testa ricorrenti,
perdita di appetito, vomito e convulsioni. Gli individui più colpiti
sperimentano episodi di simil-ictus che iniziano prima dei 40 anni Questi
episodi spesso comportano debolezza temporanea muscolare su un lato del corpo
(emiparesi), alterazione della coscienza, alterazioni della visione,
convulsioni, e forti mal di testa che assomigliano emicranie. Episodi di
simil-ictus ripetuti possono progressivamente danneggiare il cervello, con
conseguente perdita della vista, problemi di movimento, e una perdita della
funzione intellettuale (demenza). Gli episodi di simil-ictus può essere
mis-diagnosi di epilessia da un medico non è a conoscenza della condizione
MELAS.
La maggior parte delle persone affette da MELAS hanno un
accumulo di acido lattico nei loro corpi, una condizione chiamataacidosi lattica . Aumento
acidità nel sangue può portare a vomito, dolore addominale, stanchezza estrema
(stanchezza), debolezza muscolare, perdita di controllo dell’intestino, e
difficoltà di respirazione. Meno comunemente, le persone con MELAS possono
sperimentare spasmi involontari muscolari (mioclono), muscolare alterata
coordinazione ( atassia ), perdita di udito,
problemi cardiaci e renali, diabete, epilessia, e gli squilibri ormonali.
La
presentazione di alcuni casi è simile a quello della sindrome di Kearns-Sayre . [ 4 ]
La diagnosi di sindrome di MELAS si basa
sulla presentazione e il cervello di imaging clinico. La risonanza
magnetica può rivelare numerose lesioni iperintense in T2 nella materia bianca
e grigia cerebrale, mentre la tomografia computerizzata mostra atrofia
cerebrale e calcificazioni dei gangli della base. Essi mostrano che le
lesioni non sono confinate ai territori vascolari e quindi che gli episodi
acuti non sono tratti tipici. Accumulo anomalo di lattato è frequente nel
sangue e quasi costante nel liquido cerebrospinale. La biopsia muscolare è
anormale in circa l’85% dei pazienti. Essa mostra proliferazione anormale
mitocondriale (fibre rosse sfilacciate) e fibre muscolari con un difetto di
citocromo c ossidasi. Analisi del muscolo attività della catena
respiratoria può rivelare carenza del complesso o un deficit combinato dei
complessi I e IV. Identificazione della mutazione causale deve tenere
conto della costante eteroplasmia cioè la sua coesistenza con una popolazione
residuale di tipo selvaggio DNA mitocondriale. Proporzioni mutazione può
variano notevolmente tra i tessuti, ma è il più delle volte molto elevata
(superiore al 90%) e possono quindi essere studiati nel sangue. La consulenza
genetica è molto arduo nella sindrome MELAS causa della
eteroplasmia. Mutazioni del DNA mitocondriale sono trasmessi secondo
ereditarietà materna. Un uomo affetto non può trasmettere la
malattia. La mutazione verrà trasmessa lungo la linea materna, ma la sua
quota è essenzialmente imprevedibile. Anche se le proporzioni più elevate
di mutazione nel sangue del risultato madre in un rischio maggiore di avere un
bambino con grave fenotipo, ci sono molti esempi di estrema segregazione della
mutazione da madre a figlio, che impediscono la consulenza genetica efficiente
a livello individuale. L’eterogeneità possibilità nella proporzione della
mutazione tra i tessuti ostacola teoricamente diagnosi prenatale. Pochissimi
studi clinici adeguati sono stati condotti con MELAS pazienti. Un recente
ha trovato dicloroacetato ad avere effetti negativi nel medio
termine. Evoluzione spontanea della malattia con crisi acute, la
remissione e ricorrenza rende difficile valutare il miglioramento clinico
riportato in alcuni pazienti trattati con MELAS trattamenti di supporto (tra
cui il coenzima Q10 e il suo analogo idebenone, la creatina monoidrato e
arginina) o l’impatto deleterio di trattamento, come acido valproico (un
farmaco antiepilettico riferito di provocare episodi di simil-ictus). La
prognosi è scarsa. I pazienti possono morire durante un episodio
simil-ictus e, insieme a episodi ricorrenti, spesso sviluppano deterioramento
mentale, perdita della vista e dell’udito, nonché grave miopatia, che potrebbe
condurre alla perdita di autonomia.
Revisore esperto (s)Dr Anne LOMBES Ultimo aggiornamento: Luglio 2006
Genetica

La biopsia muscolare di una persona con diagnosi di MELAS ma
portare nessuna mutazione conosciuta.(A) Modificato colorazione tricromica
Gomori mostra diverse fibre rosse
sfilacciate (punta di freccia). (B) II fibre, fibre scure e qualche fibre
con collezioni anomali di mitocondri (freccia) macchia citocromo c ossidasi
mostra Type-1 leggermente macchiato e Type. Nota fibre negative citocromo
c ossidasi, come di solito visto in encefalopatia mitocondriale, acidosi
lattica e episodi di simil-ictus (MELAS). (C) succinato deidrogenasi
colorazione mostra alcuni blu fibre laceri e intensa colorazione nei mitocondri
dei vasi sanguigni (freccia). Microscopia (d) Electron mostrando raccolta
anomala di mitocondri con inclusioni paracristalline (punta di freccia), inclusioni
osmiophilic (grande punta di freccia) e vacuoli mitocondriali (piccola punta di
freccia). Abu-Amero et al. Journal of Medical Case Reports
2009. [ 5 ]
MELAS è
causata da mutazioni nei geni nel DNA mitocondriale.
NADH deidrogenasi
Alcuni dei geni ( MT-ND1 , MT-ND5 ) colpiti in MELAS
codificano proteine che sono parte della NADH deidrogenasi (chiamato
anche complesso I) in mitocondri, che aiuta a convertire l’ossigeno e zuccheri
semplici di energia.
RNA di trasferimento
Altri geni
( MT-TH , MT-TL1 , e MT-TV ) codificano mitocondriali RNA specifiche di trasferimento
( tRNA ).
Mutazioni in MT-TL1 causano più del 80 per
cento di tutti i casi di MELAS. Essi compromettono la capacità dei mitocondri
per produrre le proteine, usa l’ossigeno, e producono energia. I
ricercatori non hanno determinato come i cambiamenti nel portare DNA
mitocondriale ai segni e sintomi di MELAS specifici. Essi continuano a
studiare gli effetti di mutazioni geniche mitocondriali in diversi tessuti, in
particolare nel cervello.
La malattia è causata da mutazioni del DNA
mitocondriale. Almeno 10 diverse mutazioni sono state identificate ma 80%
dei casi sono dovuti alla 3243A> G mutazione nel gene transfer leucina RNA
( tRNA Leu ). Questa mutazione è quindi spesso definito
come la mutazione MELAS nonostante la sua associazione con diverse
presentazioni cliniche: la sua prevalenza nella popolazione generale europea è
stata stimata in 1/6250. La mutazione 3271T> C nel gene tRNA Leu è associata
con la sindrome in un ulteriore 7,5% dei pazienti
Inheritance
Questa condizione è ereditata in un modello mitocondriale, che è
anche conosciuto come eredità
materna e eteroplasmia . Questo modello di ereditarietà applica ai geni
contenuti nel DNA mitocondriale. Poiché le cellule uovo, ma non le cellule
spermatiche, contribuiscono mitocondri nello sviluppo embrionale, solo le
femmine passano condizioni mitocondriali ai loro figli.Malattie mitocondriali
possono apparire in ogni generazione di una famiglia e può colpire sia i maschi
che le femmine, ma i padri non passare tratti mitocondriali ai loro
figli. Nella maggior parte dei casi, le persone con MELAS ereditano un
gene mitocondriale alterato dalla madre. Meno frequentemente, i risultati
di disturbo una nuova mutazione in un gene mitocondriale e si verifica nelle
persone con una storia familiare di MELAS.
Prognosi
Non vi è alcun trattamento conosciuto per la malattia di base,
che è progressiva e fatale. I pazienti sono gestiti secondo quali aree del
corpo sono interessate in un momento particolare. Gli
enzimi , aminoacidi , antiossidanti e vitamine sono stati
utilizzati, ma non ci sono stati successi consistenti segnalati.
Anche se non ci sono stati studi controllati sui benefici a
lungo termine di manipolazioni dietetiche, i seguenti supplementi hanno
mostrato risultati promettenti e dato speranza ai pazienti MELAS.
·
CoQ10 è stato utile per alcuni pazienti MELAS. [ 6 ] Nicotinamide è stata utilizzata
perché complesso l accetta elettroni da NADH e infine trasferisce elettroni
CoQ10.
·
Riboflavina è stato segnalato per migliorare la funzione di un
paziente con deficit di l complesso e la mutazione 3250T-C. [ 7 ]
·
La somministrazione di L-arginina nei periodi acuti e
interictali può rappresentare una nuova potenziale terapia per questa sindrome
per ridurre i danni cerebrali a causa di compromissione della vasodilatazione
nelle arterie cerebrali a causa di ossido nitrico esaurimento. [ 8 ] [ 9 ]
·
C’è anche un caso in cui è stato utilizzato con successo
succinato per trattamento di convulsioni non controllate in MELAS pazienti,
anche se questa modalità di trattamento è ancora da indagare a fondo e
ampiamente raccomandato.
·
La diagnosi di sindrome di MELAS si basa
sulla presentazione e il cervello di imaging clinico. La risonanza
magnetica può rivelare numerose lesioni iperintense in T2 nella materia bianca
e grigia cerebrale, mentre la tomografia computerizzata mostra atrofia
cerebrale e calcificazioni dei gangli della base. Essi mostrano che le
lesioni non sono confinate ai territori vascolari e quindi che gli episodi
acuti non sono tratti tipici. Accumulo anomalo di lattato è frequente nel
sangue e quasi costante nel liquido cerebrospinale. La biopsia muscolare è
anormale in circa l’85% dei pazienti. Essa mostra proliferazione anormale
mitocondriale (fibre rosse sfilacciate) e fibre muscolari con un difetto di
citocromo c ossidasi. Analisi del muscolo attività della catena
respiratoria può rivelare carenza del complesso o un deficit combinato dei
complessi I e IV. Identificazione della mutazione causale deve tenere
conto della costante eteroplasmia cioè la sua coesistenza con una popolazione
residuale di tipo selvaggio DNA mitocondriale. Proporzioni mutazione può
variano notevolmente tra i tessuti, ma è il più delle volte molto elevata
(superiore al 90%) e possono quindi essere studiati nel sangue. La
consulenza genetica è molto arduo nella sindrome MELAS causa della
eteroplasmia. Mutazioni del DNA mitocondriale sono trasmessi secondo
ereditarietà materna. Un uomo affetto non può trasmettere la malattia. La
mutazione verrà trasmessa lungo la linea materna, ma la sua quota è
essenzialmente imprevedibile. Anche se le proporzioni più elevate di
mutazione nel sangue del risultato madre in un rischio maggiore di avere un
bambino con grave fenotipo, ci sono molti esempi di estrema segregazione della
mutazione da madre a figlio, che impediscono la consulenza genetica efficiente
a livello individuale. L’eterogeneità possibilità nella proporzione della
mutazione tra i tessuti ostacola teoricamente diagnosi prenatale. Pochissimi
studi clinici adeguati sono stati condotti con MELAS pazienti. Un recente
ha trovato dicloroacetato ad avere effetti negativi nel medio
termine. Evoluzione spontanea della malattia con crisi acute, la
remissione e ricorrenza rende difficile valutare il miglioramento clinico
riportato in alcuni pazienti trattati con MELAS trattamenti di supporto (tra
cui il coenzima Q10 e il suo analogo idebenone, la creatina monoidrato e
arginina) o l’impatto deleterio di trattamento, come acido valproico (un
farmaco antiepilettico riferito di provocare episodi di simil-ictus). La
prognosi è scarsa. I pazienti possono morire durante un episodio
simil-ictus e, insieme a episodi ricorrenti, spesso sviluppano deterioramento
mentale, perdita della vista e dell’udito, nonché grave miopatia, che potrebbe
condurre alla perdita di autonomia”Revisore esperto (s)Dr Anne LOMBES Ultimo aggiornamento: Luglio 2006
References[edit]
1. Jump up^ Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland
LP (1984). "Mitochondrial myopathy, encephalopathy, lactic acidosis, and
strokelike episodes: a distinctive clinical syndrome". Ann Neurol 16 (4):
481–8.doi:10.1002/ana.410160409. PMID 6093682.
2. Jump up^ Hirano M, Pavlakis SG (1994). "Mitochondrial
myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS):
current concepts". J Child Neurol 9 (1): 4–13.doi:10.1177/088307389400900102. PMID 8151079.
3. Jump up^ MELAS syndrome at NLM Genetics
Home Reference
4. Jump up^ Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis
SG, DeVivo DC, DiMauro S, Rowland LP (1992). "Melas: an original case and
clinical criteria for diagnosis". Neuromuscul
Disord 2 (2): 125–35. doi:10.1016/0960-8966(92)90045-8.PMID 1422200.
5. Jump up^ Abu-Amero KK, Al-Dhalaan H, Bohlega S, Hellani A, Taylor
RW (2009). "A patient with typical clinical
features of mitochondrial encephalopathy, lactic acidosis and stroke-like
episodes (MELAS) but without an obvious genetic cause: a case report". J Med Case Reports 3:
77. doi:10.1186/1752-1947-3-77. PMC 2783076. PMID 19946553.
6. Jump up^ Rodriguez MC, MacDonald JR, Mahoney DJ, Parise G, Beal MF,
Tarnopolsky MA (2007). "Beneficial effects of creatine, CoQ10, and lipoic
acid in mitochondrial disorders". Muscle
Nerve35 (2):
235–42. doi:10.1002/mus.20688. PMID 17080429.
7. Jump up^ Ogle RF, Christodoulou J, Fagan E, et al. (1997).
"Mitochondrial myopathy with tRNA(Leu(UUR)) mutation and complex I
deficiency responsive to riboflavin". J. Pediatr. 130 (1):
138–45. doi:10.1016/S0022-3476(97)70323-8.PMID 9003864.
8. Jump up^ Koga Y, Akita Y, Nishioka J, et al. (2007).
"MELAS and L-arginine therapy". Mitochondrion 7 (1-2):
133–9.doi:10.1016/j.mito.2006.11.006. PMID 17276739.
9. Jump up^ Hirata K, Akita Y, Povalko N, et al. (2007).
"Effect of l-arginine on synaptosomal mitochondrial function". Brain
Dev 30 (4): 238–
MELAS APPROFONDIMENTO medscape
La sindrome MELAS (encefalomiopatia mitocondriale con acidosi lattica e episodi
simili a ictus) è una patologia progressiva caratterizzata da disturbi
neurologici acuti paragonabili a ischemie cerebrali, associati a iperlactatemia
e miopatia mitocondriale. La prevalenza è sconosciuta. Durante l’infanzia o la
prima adolescenza, i pazienti sono soggetti di solito a crisi acute, forse
causate da un’infezione o da uno sforzo fisico. Queste crisi si associano a
cefalee, vomito e a volte ad episodi pseudoischemici, emiparesi e emianopsia.
Si manifestano spesso in pazienti con sintomi cronici come deficit motorio,
sordità, diabete, bassa statura, cardiomiopatia, ritardo dello sviluppo,
difficoltà di apprendimento, di memoria e di attenzione. La malattia è dovuta
alle mutazioni del DNA mitocondriale. Sono state identificate 10 mutazioni
differenti ma l’80% dei casi è dovuto alla mutazione 3243A>G nel gene del
tRNA della leucina (tRNA Leu). Questa mutazione viene definita spesso mutazione
MELAS, sebbene sia associata a segni clinici diversi; la sua prevalenza in
Europa è stimata in 1 su 6250 e si associa alla sindrome in oltre il 7,5% dei
pazienti. La diagnosi si basa sulle manifestazioni cliniche e sulla risonanza
magnetica cerebrale. La risonanza magnetica può rivelare la presenza di
numerose lesioni profonde nella materia grigia e bianca del cervello, mentre la
TAC identifica atrofia cerebrale e calcificazioni dei gangli basali. La
risonanza e la TAC mostrano che le lesioni non sono confinate nelle aree
vascolari e di conseguenza gli episodi acuti non si possono considerare tipici
ictus. La concentrazione anomala di lattato è frequente nel sangue e pressoché
costante nel liquido cerebrospinale. La biopsia muscolare è anomala nell’85%
dei pazienti, mostra una proliferazione mitocondriale atipica (fibre rosse
lacerate) e fibre muscolari con deficit di citocromo-ossidasi. L’analisi delle
attività enzimatiche della catena respiratoria muscolare può rivelare un
deficit del complesso I o un deficit combinato dei complessi I e IV.
L’identificazione della mutazione causale deve tenere conto della sua costante
eteroplasmia, cioè la sua coesistenza con una popolazione residuale di DNA
mitocondriale normale. La proporzione delle mutazioni varia notevolmente a
seconda dei tessuti, ma spesso può essere molto elevata (oltre il 90% della
popolazione del DNA mitocondriale) e può essere ricercata anche nel sangue. La
terapia è complicata dall’eteroplasmia. La mutazione è trasmessa per eredità
materna. Un maschio affetto non è in grado di trasmettere la malattia. Sebbene
una proporzione elevata della mutazione nel sangue della madre aumenti il
rischio di nascita di un bambino gravemente affetto dalla malattia, esistono
molti esempi di segregazione estrema madre-figlio, che rendono difficile la
consulenza genetica. La presenza di percentuali eterogenee della mutazione nei
diversi tessuti impedisce, in teoria, la diagnosi prenatale. Sono state
effettuate poche sperimentazioni cliniche. Una sperimentazione recente ha scoperto
che il dicloroacetato ha un effetto deleterio nel medio termine. L’evoluzione
spontanea, fatta di crisi seguite da recuperi e ricadute, rende difficile
valutare il miglioramento in alcuni pazienti sottoposti a trattamenti (che
utilizzano il coenzima Q10 e l’analogo idebenone, la creatina monoidrato e
l’arginina) o il peggioramento dovuto ad alcuni trattamenti come l’acido
valproico (farmaco a effetto antiepilettico responsabile di episodi simili a
ictus). La prognosi è grave. Gli episodi potrebbero provocare il decesso del
paziente e la loro ricorrenza nel lungo periodo può causare un deterioramento
mentale, perdita della vista e dell’udito e grave miopatia, che potenzialmente
può contribuire alla perdita dell’autonomia.
Sfondo
Miopatia mitocondriale,
encefalopatia, acidosi lattica, e ictus (MELAS) La sindrome è una malattia
neurodegenerativa progressiva. I pazienti possono presentare
sporadicamente o come membri di pedigree materni con un’ampia varietà di
presentazioni cliniche. La presentazione tipica dei pazienti con sindrome
MELAS include caratteristiche che compongono il nome del disturbo, come
encefalomiopatia mitocondriale, acidosi lattica , ed
episodi di. Altre caratteristiche, come le convulsioni, diabete mellito , perdita
dell’udito, malattie cardiache, bassa statura ,
endocrinopatie, intolleranza all’esercizio, e disfunzioni neuropsichiatrici
sono chiaramente parte del disordine.
Fisiopatologia
Episodi di ictus e miopatia
mitocondriale caratterizzano la sindrome MELAS. Coinvolgimento organo multisistemica
è visto, compreso il SNC, muscolo scheletrico, occhio, muscolo cardiaco, e, più
raramente, la GI e renale. [1]
Circa l’80% dei pazienti con le
caratteristiche cliniche della sindrome MELAS hanno un eteroplasmica mutazione
puntiforme A-to-G nel ciclo dihydrouridine del RNA di trasferimento (tRNA) Leu
(UUR) gene alla coppia di basi (bp) 3243 (vale a dire, 3243 A
→ G mutazione). [2] Tuttavia, altri DNA
mitocondriale (mtDNA) si osservano, tra cui il m.3244 G → A, m.3258 T
→ C, m.3271 T → C, e m.3291 T → C nel mitocondriale tRNA Leu
(UUR) gene.
La patogenesi degli episodi di
simil-ictus nella sindrome MELAS non è stato completamente chiarito. [3] Questi episodi ictus
metaboliche può essere non vascolari e grazie alla transitoria fosforilazione
ossidativa (OXPHOS) disfunzioni all’interno del parenchima cerebrale. Un
angiopatia mitocondriale di piccoli vasi è responsabile per migliorare il
contrasto delle regioni colpite e alterazioni mitocondriali di cellule
endoteliali e cellule muscolari lisce dei vasi sanguigni. La disfunzione
multisistemica in pazienti con sindrome MELAS può essere dovuta sia
parenchimale e difetti OXPHOS vascolari. Aumento della produzione di
radicali liberi in associazione con un difetto OXPHOS che porta a
vasocostrizione può compensare l’effetto di potenti vasodilatatori (ad esempio,
ossido nitrico).
Gli episodi di simil-ictus
inusuali e maggiore morbilità osservata nella sindrome di MELAS può essere
secondaria ad alterazioni dell’omeostasi ossido di azoto che causano danni
microvascolari. L’ossido nitrico può legare i citocromo c ossidasi-positive
siti nei vasi presenti nel SNC sangue, spostando ossigeno eme-legato e
conseguente diminuita disponibilità di ossigeno nel tessuto circostante e
diminuito ossido nitrico libero. Inoltre, l’accoppiamento della
disfunzione mitocondriale vascolare con depressione corticale diffusione
potrebbe essere alla base della distribuzione selettiva di lesioni ischemiche
nella corteccia posteriore in questi soggetti.
Mutazioni in questo disturbo
influenzano la funzione del tRNA mitocondriale, che porta alla rottura del processo
globale di intramitocondriale sintesi proteica. Le misurazioni di attività
enzimatiche respiratorie nei mitocondri intatti hanno rivelato che più della
metà dei pazienti con sindrome di MELAS può avere l’I complessi o complesso I +
carenza IV. Un primo rapporto è apparente tra MELAS e carenza del
complesso. La sintesi proteica è diminuito può infine portare alla
diminuzione osservata dell’attività della catena respiratoria da traduzione
ridotto di geni UUG-ricchi come ND6 (componente del
complesso). [4]
Inoltre, studi hanno rivelato che
il 3243 A → G mutazione produce un grave difetto catena respiratoria
combinato in mioblasti, con quasi totale assenza di assemblaggio del complesso
I, IV e V, e una leggera diminuzione del complesso assemblato III. Questo
difetto di assemblaggio avviene nonostante una modesta riduzione del tasso
globale di sintesi delle proteine mitocondriali. Traduzione
di alcuni polipeptidi è diminuita, e le prove di aminoacidi misincorporation è
notato in altri.
Epidemiologia
Frequenza
Stati Uniti
Nessun stime relative alla
prevalenza della mutazione MELAS comuni sono disponibili per la popolazione del
Nord America; tuttavia, la sindrome è stata osservata essere meno frequente
nei neri.
Internazionale
La prima valutazione della
epidemiologia dei disturbi mitocondriali trovato una prevalenza di oltre il
10,2 per 100.000 per la mutazione m.3243A → G nella popolazione
finlandese adulta. Se il presupposto è fatto che tutti i parenti materni
di primo grado di un portatore di mutazione verificato porto anche la
mutazione, prevalenza aumenta a più di 16,3 per 100.000. Questa alta
prevalenza suggerisce che i disordini mitocondriali possono costituire una
delle maggiori categorie diagnostiche di malattie neurogenetiche tra gli
adulti. In Inghilterra del Nord, la prevalenza di questa mutazione nella
popolazione adulta è stato determinato in circa 1 ogni 13.000.
Mortalità /
morbilità
La malattia progressiva ha una
morbilità e mortalità. Il Encefalomiopatia, associata a episodi di ictus
seguiti da emiplegia e emianopsia, è grave. Convulsioni focali e generali
possono verificarsi in associazione con questi episodi.
Altre anomalie che possono essere
osservati sono dilatazione ventricolare, atrofia corticale, e gangli basali
calcificazione. Deterioramento mentale di solito progredisce attacchi
episodici dopo ripetuti. Anomalie psichiatrici e declino cognitivo (ad
esempio, alterazione dello stato mentale, schizofrenia ) possono accompagnare
gli episodi di simil-ictus. disturbo bipolare è un’altra
anomalia psichiatrica osservata nella sindrome MELAS. disturbi dello spettro autistico (ASD), con o senza ulteriori funzioni neurologiche possono essere
prime presentazioni del m 0,3243 A → G mutazione. Miopatia può
essere debilitante.L’encefalopatia può progredire verso la
demenza; infine, il decorso clinico declina rapidamente, portando a grave
disabilità e morte prematura.
Un’altra causa di elevata
mortalità è la caratteristica meno comune di coinvolgimento cardiaco, che
possono includere cardiomiopatia ipertrofica ,ipertensione e anomalie di conduzione,
come blocchi atrio-ventricolare, sindrome del QT lungo , o sindrome di Wolff-Parkinson-White . Sono stati trovati soggetti con sindrome di MELAS
aver aumentato crescente rigidità aortica e dell’aorta dimensioni ingrandite
suggerendo rimodellamento vascolare. Dissezione aortica radice è stata
trovata in un paziente con la sindrome MELAS. [5] Alcuni pazienti
possono sviluppare la sindrome di Leigh (cioè, subacuta necrotizzante
encefalopatia). I pazienti possono sviluppare insufficienza renale a causa
di glomerulosclerosi focale segmentale.
Più raramente, questi pazienti
possono presentare gravi disturbi della motilità gastrointestinale e
disfunzione endocrina, compreso l’ipotiroidismo el’ipertiroidismo .
Gara
Nessuna predilezione per un
particolare gruppo etnico è notato.
Sesso
Nessuna predilezione sessuale è
presente.
Età
In molti pazienti con sindrome di
MELAS, la presentazione avviene con il primo episodio di ictus, di solito
quando un individuo è affinato 4-15 anni. Meno spesso, l’esordio della
malattia può avvenire nell’infanzia con tappe dello sviluppo in ritardo e
difficoltà di apprendimento. Una presentazione del disturbo è stato
segnalato in un neonato di 4 mesi.
Storia
Insorgenza del disturbo può
essere miopatica con debolezza, facile affaticamento, e intolleranza
all’esercizio.
Miopatia mitocondriale,
encefalopatia, acidosi lattica, e ictus (MELAS) sindrome esordio possono
verificarsi nella prima infanzia con una storia di ritardo dello sviluppo e
difficoltà di apprendimento. Il ritardo dello sviluppo, difficoltà di
apprendimento, o disturbo da deficit di attenzione si trova principalmente in pazienti prima dello sviluppo del
primo colpo. Un quadro encefalopatica che è progressiva e porta a demenza
può essere presente. I pazienti possono essere apatico. Il livello di
funzionamento cognitivo peggiora nel corso del tempo in base al punteggio
Karnofsky in pazienti completamente sintomatici.
Ritardo di crescita può essere la funzione di presentare in alcuni pazienti con
sindrome MELAS.
Episodi di simil-ictus sono la
caratteristica caratteristica di questo disturbo.Inizialmente, gli episodi
possono manifestarsi con vomito e mal di testa che può durare diversi
giorni. Questi pazienti possono anche sperimentare episodi di crisi
epilettiche e anomalie visive seguiti da emiplegia. Tipi di crisi possono
essere tonico-cloniche o mioclonica.
Emicrania mal di testa o
migrainelike osservati in questi pazienti possono anche riflettere gli episodi
di simil-ictus. Pedigree dei pazienti con sindrome MELAS classica
identificare molti membri le cui manifestazioni sono solo mal di testa.
I pazienti possono avere
lamentele visivi a causa di oftalmoplegia, e possono sperimentare la cecità a
causa di atrofia ottica e le difficoltà con visione notturna a causa della
retinite pigmentosa.
Alcuni pazienti possono avere
perdita di udito, che può accompagnare il diabete. Si può osservare in
associazione con il classico disturbo della sindrome MELAS. [8]
Polidipsia e poliuria possono
essere i segni che presentano del diabete; diabete sembra essere la
manifestazione più comune della sindrome MELAS. Di solito, il diabete di
tipo 2 è descritto in individui con sindrome di MELAS, anche se di tipo 1
(precedentemente chiamato diabete insulino-dipendente), può anche essere
osservato. Linee guida per la diagnosi e la gestione di tipo sono state
stabilite diabete 2. [9]
Palpitazioni e mancanza di
respiro possono essere presenti in alcuni pazienti con sindrome MELAS
secondaria ad anomalie della conduzione cardiaca, come la sindrome di Wolff-Parkinson-White. I
pazienti possono sperimentare mancanza di respiro secondaria a cardiomiopatia,
che di solito è di tipo ipertrofico; tuttavia,cardiomiopatia dilatativa è stato anche descritto.
Insorgenza acuta di
manifestazioni gastrointestinali (ad esempio, insorgenza acuta di dolore
addominale) può riflettere pancreatite , colite ischemica, e ostruzione intestinale. [10]
Intorpidimento, formicolio e
dolore alle estremità possono essere manifestazioni di neuropatia periferica.
Disturbi psichiatrici (ad
esempio, depressione, disturbo bipolare), sono stati associati con il m.3243 A
→ G mutazione. La demenza è stata un’altra manifestazione
clinica. Inoltre, i disturbi dello spettro autistico (ASD) sono stati
associati con il 3243 A → G mutazione.
I pazienti possono sviluppare
caratteristiche di ipotiroidismo e ipertiroidismo
Alcuni pazienti possono
sviluppare apnea e un’andatura atassica in associazione con le caratteristiche
neuroradiologiche della sindrome MELAS.
Oliguria possono essere associati
con la sindrome MELAS e può indicare l’insorgenza della sindrome nefrosica .
I pazienti con sindrome di MELAS
possono avere coinvolgimento vascolare funzionale. Aortica dissezione
radicale è stato riportato in un paziente con la sindrome MELAS.
Esame Fisico
I pazienti con sindrome di MELAS
possono presentare ipertensione.
Miopatia presenta con ipotonia e
debolezza. Muscoli prossimali tendono ad essere più coinvolti di muscoli
distali. Muscolatura è sottile, ei pazienti possono presentare con un
volto miopatico.
Episodi di simil-ictus possono
presentare convulsioni, anomalie visive, intorpidimento, emiplegia, e
l’afasia. [11] episodi può essere seguita da emiplegia transitoria o
emianopsia, che dura un paio d’ore a diverse settimane. Altre
caratteristiche di esame neurologico possono includere atassia, tremore,
mioclono, distonia, disturbi visivi, e la cecità corticale. Alcuni
pazienti possono presentare oftalmoplegia e ptosi.
In esame oftalmologico, i
pazienti hanno presentato con retinite pigmentosa.
La sordità neurosensoriale è
stato segnalato come parte della malattia in circa il 25% dei pazienti con
sindrome MELAS.
Cardiomiopatia con segni di insufficienza cardiaca congestizia (CHF) può anche essere osservato dopo un esame fisico. [12]
Manifestazioni cutanee di porpora
cutanea, irsutismo, e squamosa, pruriginosa, eritema diffuso con reticolare
pigmentazione possono essere osservati nei pazienti con sindrome MELAS.
Bassa statura può essere la prima
manifestazione della sindrome MELAS in molti pazienti.
Cause
Sindrome MELAS è stata associata
con almeno 6 mutazioni puntiformi differenti, di cui 4 si trovano nello stesso
gene, il tRNA Leu (UUR) gene. La
mutazione più comune, che si trova nel 80% degli individui con sindrome di
MELAS, è una transizione A → G al nucleotide (nt) 3243 nel tRNA Leu
(UUR) gene. Un ulteriore 7,5% ha un eteroplasmica mutazione
puntiforme T → C a 3271 bp nella coppia nucleotide terminale del gambo
anticodone del tRNA Leu (UUR) gene. Inoltre,
un fenotipo MELAS è stato osservato associato ad un m.13513G → Una
mutazione nel ND5 gene e in carenza di POLG.
Queste mutazioni sono
eteroplasmica, che riflette le diverse percentuali di mutato mtDNA presenti in
diversi tessuti. Eteroplasmia variabile tra gli individui affetti da
sindrome di MELAS riflette segregazione variabile l’ovulo. Mutazioni in
tRNA Lys si può aspettare di avere un effetto importante sulla traduzione e la
sintesi proteica nei mitocondri. Il mitocondriale umano MELAS disturbo
associata tRNA Leu (UUR)mutazione causa la carenza
aminoacilazione e un difetto concomitante inizio della traduzione.
Omeostasi del calcio anormale
conseguente danno neuronale è stato suggerito come un altro meccanismo che
contribuisce al coinvolgimento CNS osservata nella sindrome MELAS.
I pazienti con sindrome MELAS
sono stati trovati per avere una marcata diminuzione dell’attività del
complesso I. Gli effetti principali osservati secondaria a nt 3243 e 3271 nt
mutazioni sono state una riduzione della sintesi proteica e l’attività del
complesso I. Questi effetti sono stati dimostrati attraverso cibridi che
studiano in cui le linee cellulari umane senza mtDNA si fondono con mitocondri
esogeni contenenti 0-100% della mutazione m.3243 comune. Cybrids con più
del 95% di DNA mutante era diminuita velocità di sintesi delle proteine
mitocondriali, portando a difetti della catena respiratoria.
Procedere
alla Diagnosi differenziale
Diagnosi differenziale
·
Sindrome da anticorpi antifosfolipidi
·
Blocco atrioventricolare, Second Degree
·
Blocco atrioventricolare, Terzo Grado,
acquisita
·
Lunga catena acil CoA deidrogenasi,
deficit di
·
A catena media acil-CoA deidrogenasi
·
Mitocondriale
DNA polimerasi carenza (POLG)
·
Disturbo dell’Umore: disturbo bipolare
·
Disturbo dell’Umore: Depressione
·
Oliguria
·
Pancreatite e pseudocisti pancreatica
·
Tachicardia sopraventricolare, sindrome
di Wolff-Parkinson-White
Studi di laboratorio
I seguenti studi sono indicati
nei pazienti con miopatia mitocondriale, encefalopatia, acidosi lattica, e
ictus (MELAS) sindrome:
·
Acido siero lattico, acido piruvico siero, liquido cerebrospinale
(CSF) di acido lattico e acido piruvico CSF
o
L’acidosi lattica è una caratteristica importante della sindrome
MELAS.Vedere l’immagine qui sotto per la classificazione fisiopatologico di
acidosi lattica.
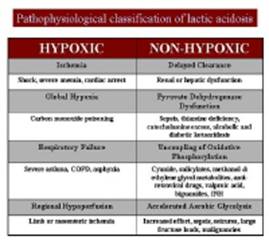
classificazione fisiopatologico di acidosi
lattica.
o
In generale, acidosi lattica non porta ad acidosi metabolica
sistemica, e può essere assente nei pazienti con notevole coinvolgimento del
SNC.
o
In alcuni individui con sindrome di MELAS, livelli di acido
lattico possono essere normali nel sangue, ma elevata in CSF.
o
In difetti della catena respiratoria, il rapporto tra lattato e
piruvato è alta.
·
Livelli di creatina chinasi sierici
o
I livelli sierici di creatina chinasi sono leggermente ad
aumentare moderatamente in alcuni pazienti con sindrome MELAS.
o
Livelli tendono ad aumentare durante e immediatamente dopo
episodi.
·
Respiratori attività degli enzimi della catena nel muscolo
scheletrico
o
Se una biopsia muscolare viene eseguita per perseguire una
valutazione diagnostica, quindi verificare le attività degli enzimi della
catena respiratoria.
o
I pazienti con sindrome di MELAS sono stati trovati ad avere
segnato la carenza di attività complesso della catena respiratoria I.
o
Alcuni pazienti con la malattia hanno un deficit combinato del
complesso I e IV complessa.
·
Mitocondriale analisi della mutazione del DNA sul sangue, muscolo
scheletrico, follicoli dei capelli, mucosa orale, e sedimento urinario
o
Individui con più gravi manifestazioni cliniche della sindrome
MELAS hanno generalmente superiore al 80% mtDNA mutante nei tessuti stabili
come muscolare.
o
In cellule in rapida divisione, come i componenti delle linee
ematopoietiche, la m.3243 A → G mutazione può separare a livelli
estremamente bassi, rendendo la diagnosi genetica dal sangue difficile.
o
La percentuale della mutazione diminuisce progressivamente in DNA
isolato dal sangue. Il carico mutante isolata dal sangue non è né utile
per la prognosi né per la valutazione funzionale.
o
Sedimento urinario, seguita da fibroblasti della pelle e mucosa
orale, sono i tessuti accessibili della scelta, perché sono di facile accesso e
il carico di mutazione è maggiore di quella riscontrata nel sangue.
o
Se la diagnosi si sospetta ancora dopo normali risultati delle
analisi di mutazione del mtDNA in questi tessuti, una biopsia muscolare
scheletrico è necessario per confermare o escludere la presenza della
mutazione.
Studi di imaging
I seguenti studi di imaging
possono essere presentate:
·
TAC o risonanza magnetica del cervello
o
TAC o risonanza magnetica del cervello a seguito di un episodio di
ictus rivela un lucency coerente con infarto.
o
Più tardi, atrofia cerebrale e calcificazioni possono essere
osservati in studi di brain imaging.
o
I pazienti con sindrome di MELAS che hanno una presentazione
simile alla sindrome di Leigh possono avere calcificazioni nei gangli basali.
·
Tomografia ad emissione di positroni (PET) studi
o
Gli studi PET possono rivelare un tasso metabolico cerebrale
ridotta per l’ossigeno.
o
Aumento flusso sanguigno cerebrale nelle regioni corticali può
osservare.
o
PET può dimostrare di mantenimento del tasso metabolico cerebrale
per il glucosio.
·
Singolo fotone studi CT emissione
o
Emissione di singoli fotoni tomografia computerizzata (SPECT)
studi possono accertare colpi in individui con sindrome MELAS con un
tracciante, N -isopropyl-p- [123-I] -iodoamphetamine.
o
Il tracciante accumula nella regione parietooccipital, e può
delineare l’estensione della lesione. Studi SPECT vengono utilizzati per
monitorare l’evoluzione della malattia.
·
Spettroscopia di risonanza magnetica protonica ( 1 H-MRS):
questo viene utilizzato per identificare anomalie metaboliche, tra cui il
rapporto lattato-to-creatina sia muscolo o cervello e il sistema nervoso
centrale è diminuito Nrapporto -acetylaspartate-to-creatina nelle
regioni di ictus. Con questa tecnica, regioni elevati di lattato sono
stati rilevati mentre i livelli sierici sono normali.
·
Ecocardiografia: Questo è utile per valutare per cardiomiopatia
ipertrofica e dilatativa e le dimensioni della radice aortica; tuttavia,
cardiomiopatia non è una caratteristica comune nelle persone con sindrome
MELAS.
Altri test
EEG di solito sono
anormali. Scariche epilettiformi picco sono di solito presenti.
ECG viene utilizzato per cercare
anomalie di conduzione con aritmie ventricolari.ECG può identificare
coinvolgimento presintomatica cardiaco, sindromi pre-eccitazione, e blocco di
conduzione cardiaca.
Procedure
Considerare l’esecuzione di una
biopsia muscolare, se la sindrome MELAS si sospetta e se l’analisi di mutazione
del mtDNA nel sangue e altri tessuti accessibili fornisce risultati
banali. In rapida divisione linee di cellule, le mutazioni possono
separare a livelli bassi, rendendo la diagnosi genetica dal sangue difficile.
I risultati istologici
In biopsie muscolari colorate con
ematossilina eosina, la variazione si osserva nelle misure di tipo 1 e di tipo
2 in fibra, che rappresenta alterazioni miopatiche.
Fibre rosse sfilacciate sono il
segno distintivo della sindrome MELAS. Le fibre rosse sfilacciate colorano
rosso brillante con corpi citoplasmatici occasionali con colorazione
tricromica. Fibre rosse sfilacciate solito macchia positiva con macchia
citocromo ossidasi.
Colorazione con acido periodico
di Schiff, nicotinamide adenina dinucleotide (NADH) deidrogenasi tetrazolio
reduttasi, o per succinico deidrogenasi dimostra una maggiore attività
subsarcolemmale. Questa proliferazione mitocondriale è stato osservato
anche nei vasi sanguigni e viene determinato utilizzando una macchia per la
succinato deidrogenasi.
La microscopia elettronica mostra
un aumento del numero e le dimensioni dei mitocondri, alcuni con corpi
paracristalline.
Cura Medica
La valutazione per la sindrome
(MELAS) miopatia mitocondriale, encefalopatia, acidosi lattica, ed ictus può
essere eseguita su base ambulatoriale, se il paziente è stabile. La
valutazione può essere costituita da nella determinazione dei livelli di
lattato nel siero e piruvato sierico, studi sulle mutazioni del mtDNA sul
sangue, e studi di brain imaging (ad esempio, la scansione testa CT, MRI del
cervello, spettroscopia di risonanza magnetica [ 1 H-MRS]) protonica
del cervello. Può essere eseguita come procedura elettiva la biopsia muscolare
per gli enzimi mitocondriali e l’analisi di mutazione del DNA per cui il
paziente è ricoverato in ospedale.
In casi di scompenso acuto,
effettuare studi pazienti ricoverati in fase acuta e in seguito la
stabilizzazione del paziente.
Sono disponibili varie misure di
sostegno, anche se nessuno studio controllato ha dimostrato efficacia. I
benefici a lungo termine di manipolazioni dietetiche sono sconosciuti. I
miglioramenti in alcuni pazienti possono essere correlati a una migliore stato
nutrizionale e idratazione.
Sono stati utilizzati i seguenti
farmaci:
·
Il trattamento con coenzima CoQ10 è stato utile in alcuni pazienti
con sindrome MELAS. Nessun effetto negativo sono stati riportati dalla sua
amministrazione.
·
Menadione (vitamina K-3), phylloquinone (vitamina K-1), e
ascorbato sono stati utilizzati per donare elettroni citocromo c . Idebenone
è stato utilizzato anche per il trattamento di questa condizione, e sono stati
segnalati miglioramenti nella alterazioni cliniche e metaboliche.
·
Riboflavina è stato segnalato per migliorare la funzione di un
paziente con carenza del complesso e la m.3250 T → C
mutazione. Nicotinamide è stata utilizzata perché complesso accetta
elettroni da nicotinamide adenina dinucleotide (NADH) e infine trasferisce
elettroni CoQ10.
·
Dicloroacetato è un altro composto usato con questi agenti, poiché
i livelli di lattato si abbassano nel plasma e nel liquido cerebrospinale
(CSF); pazienti riferito possono rispondere in modo favorevole. La
neuropatia sensoriale può causare dopo un uso prolungato di questo farmaco.
·
Succinato di sodio è stato utilizzato, e un paziente con la
sindrome MELAS riferito avuto meno episodi di simil-ictus con il suo
uso; tuttavia, succinato di sodio non è lo standard di
cura. Ulteriori indagini è necessario.
·
Creatina monoidrato è stato utilizzato anche, e un aumento della
forza muscolare in attività anaerobiche e aerobiche ad alta intensità è stata
riportata.
·
La somministrazione di L-arginina nei periodi acuti e interictali
può rappresentare una nuova potenziale terapia per questa sindrome per ridurre
il danno cerebrale dovuto alla vasodilatazione alterata in arterie
intracerebrali causa ossido nitrico esaurimento.
Consulti
I seguenti consultazioni possono
essere presentate:
·
Genetista
·
Neurologo (per valutare il paziente per gli episodi ictus)
·
Cardiologo (per la valutazione di cardiomiopatia, aritmie e
ipertensione)
·
Nefrologo (da valutare per l’insorgenza della sindrome nefrosica)
·
Oculista (di valutare per la retinopatia pigmentosa)
·
Endocrinologo (valutare per disfunzioni endocrine come il diabete
mellito, ipotiroidismo, ipertiroidismo e ipoparatiroidismo)
·
Psichiatra (per valutare per i disturbi affettivi)
·
Neuropsicologo (valutare per disturbi dello spettro autistico
[ASD])
Dieta
L’effetto della manipolazione
alimentare non è completamente noto, e l’efficacia di integratori alimentari
non è provata. Aciduria dicarbossilici e compromissione secondaria di
acidi grassi a catena lunga ossidazione (LCFAO) possono verificarsi nei
disturbi mitocondriali. Miglioramento osservato in molti pazienti è
probabilmente legato a una migliore alimentazione.
Attività
Nei pazienti con miopatie
mitocondriali, formazione tapis roulant moderata può comportare il
miglioramento della capacità aerobica e un calo dei livelli di lattato di
riposo e postexercise lattato. Allenamento concentrico può anche giocare
un ruolo importante perché dopo un breve periodo di esercizio concentrica
formazione di un notevole incremento riferito avviene nel rapporto di
tipo-to-mutante mtDNA selvatici e della percentuale di fibre muscolari con
normale attività della catena respiratoria.
Riassunto Farmaci
Per gli individui con miopatia
mitocondriale, encefalopatia, acidosi lattica, e ictus (MELAS) sindrome e per
quelli con altri disturbi della fosforilazione ossidativa (OXPHOS), terapie
metabolici vengono somministrati per aumentare la produzione di adenosina
trifosfato (ATP) e per rallentare o arrestare il deterioramento di questa
condizione e di altri encefalomiopatie mitocondriali. Terapie metaboliche
utilizzate per la gestione della sindrome MELAS comprendono carnitina, CoQ10,
fillochinone, menadione, ascorbato (vale a dire, l’acido ascorbico),
riboflavina, nicotinamide, creatina monoidrato, idebenone, succinato, e
dicloroacetato. Tuttavia, la valutazione dell’efficacia di questi composti è
lungi dall’essere completa, ed efficacia si ritiene essere limitata ai casi
individuali.
Il trattamento con CoQ10 è stato
utile in alcuni pazienti con sindrome MELAS. Nessun effetto negativo sono stati
riportati dalla sua amministrazione. Menadione (vitamina K-3),
phylloquinone (vitamina K-1), e ascorbato sono stati utilizzati per donare
elettroni citocromo c . Idebenone è stato utilizzato
anche per il trattamento di questa condizione, e sono stati segnalati
miglioramenti nella alterazioni cliniche e metaboliche. Riboflavina è
stato segnalato per migliorare la funzione di un paziente con carenza del
complesso e la m.3250 T → C mutazione. Nicotinamide è stata
utilizzata perché complesso accetta elettroni da nicotinamide adenina dinucleotide
(NADH) e infine trasferisce elettroni Q10. Dicloroacetato è un altro
composto usato con questi agenti, perché i livelli di lattato si abbassano nel
plasma e nel liquido cerebrospinale (CSF). I pazienti riferito possono
rispondere in modo favorevole.
Un paziente con la sindrome MELAS
riferito ha avuto meno episodi di simil-ictus con l’uso di succinato di
sodio; tuttavia, succinato di sodio non è lo standard di cura, e sono
necessarie ulteriori indagini. Un aumento della forza muscolare in
attività anaerobica e aerobica ad alta intensità è stata riportata con la
somministrazione di creatina monoidrato.
La somministrazione di Arginina
nei periodi acuti e interctritici degli episodi di simil-ictus della sindrome
MELAS, può rappresentare una nuova potenziale terapia per ridurre i danni al
cervello a causa della disfunzione mitocondriale, ed è una delle terapie più
promettenti ad oggi. Sulla base l’ipotesi che gli episodi di simil-ictus
nella sindrome MELAS sono attivati da vasodilatazione alterata
nelle arterie cerebrali dovute alla diminuzione dei livelli di circolante NO,
elevazione di arginina e livelli di NO può migliorare questo
effetto. Inoltre, L-arginina può modulare l’eccitazione da
neurotrasmettitori a terminazioni nervose e tali effetti potrebbe contribuire
ad alleviare i sintomi simil-ictus nella sindrome MELAS. I pazienti con
MELAS possono avere meno probabilità di avere episodi di ictus, migliorando la
loro funzione endoteliale con la supplementazione orale di L-arginina.
Vitamine e integratori
alimentari
Riassunto Class
Le vitamine sono sostanze
organiche del corpo richiede in piccole quantità per vari processi
metabolici. Le vitamine possono essere sintetizzati in piccole o
insufficienti quantità nel corpo o no sintetizzati a tutti, richiedendo così l’integrazione. Alcuni
case report che utilizzano integratori alimentari hanno riportato un
miglioramento dei sintomi del paziente.
Visualizza informazioni complete droga
Può essere utile per il
trattamento / prevenzione di episodi di ictus nella sindrome MELAS. Gli
episodi di simil-ictus nella sindrome MELAS può essere innescato da
vasodilatazione alterata nelle arterie cerebrali dovute alla diminuzione dei
livelli circolanti di NO; quindi, elevazione di arginina e aumentata
sintesi di NO può migliorare questo effetto.
Aumenta la produzione di
ornitina, che facilita l’incorporazione di azoto rifiuti nella formazione di
citrullina e argininosuccinato. Fornisce 1 mol di urea più 1 mol ornitina
per mole di arginina quando spaccati da arginase.
Visualizza informazioni complete droga
Un derivato di aminoacido,
sintetizzato da metionina e lisina, richiesto nel metabolismo
energetico. Può promuovere l’escrezione degli acidi grassi in eccesso nei
pazienti con difetti nel metabolismo degli acidi grassi o acidopathies organici
specifici che causano esteri acil CoA di bioaccumulo.
In carenza di carnitina
secondaria associata alla sindrome MELAS, carnitina può ripristinare
generazione di liberi CoA ed evitare carnitina esaurimento. Se si verifica
la sindrome MELAS associata LCFAO difetti, uso della carnitina è discutibile
perché può aumentare la formazione di acilcarnitine a catena lunga, che possono
causare aritmogenesi ventricolare.
Visualizza informazioni complete droga
Un chinone liposolubile, la cui
funzione è trasferimento di elettroni dal complesso I al complesso
III. Sembra stabilizzarsi complessi OXPHOS situati in membrana
mitocondriale interna; può anche agire come potente antiossidante per i
radicali liberi. È stato osservato Miglioramento della debolezza muscolare
e una diminuzione di lattato sierico.
Idebenone (Avan)
I dati sono
limitati; Tuttavia, si ritiene che permettono di migliorare il metabolismo
cerebrale e migliorare la funzione di sistema elettronico di trasferimento dei
mitocondri cerebrali. Inoltre inibisce la perossidazione lipidica della
membrana mitocondriale, quindi, aumentando l’attività respiratoria
mitocondriale.
È stato usato per il trattamento
di pazienti affetti da sindrome MELAS sulla base di effetti fisiologici
proposti come antiossidante, effetto presunta sulle svalutazioni di breve
termine e memoria a lungo termine, e la somiglianza strutturale CoQ10. Non
approvato per l’uso in pazienti negli Stati Uniti; Tuttavia, è stato
utilizzato in Giappone. Miglioramento anomalie cliniche e metaboliche si
osserva nei pazienti con sindrome MELAS. Non sono noti effetti avversi.
Visualizza informazioni complete droga
Dopo la conversione di flavina
monofosfato e flavina adenina dinucleotide, funziona come cofattore per il
trasporto degli elettroni in complesso I, complesso II, e il trasferimento di
elettroni flavoproteina. Secondo quanto riferito di beneficio in caso di
carenza del complesso e MELAS.
Visualizza informazioni complete droga
Può essere utile nei pazienti
individuali come antiossidante.
Menadione
(vitamina K-3)
È stato riportato aneddoticamente
per migliorare il metabolismo del fosfato cellulare; migliora tasso di
riduzione fumarato permettendo il trasferimento di elettroni da S3 gruppo di
zolfo ferro del complesso II; sembra migliorare il trasferimento di
elettroni dopo complesso I inibizione da rotenone. Anche se il passaggio
attraverso la placenta è povero, somministrare con cautela nei pazienti in
stato di gravidanza con sindrome MELAS vicino al termine, perché emolisi e
iperbilirubinemia riferito hanno colpito i neonati.
Visualizza informazioni complete droga
Può avere effetti benefici nei
pazienti con MELAS e altri disturbi mitocondriali;effetto può essere correlato
ad un aumento della creatina intracellulare e / o contenuti fosfocreatina, che
possono essere coinvolte nel mantenimento ATP cellulare e nella stabilizzazione
della permeabilità transizione pore con conseguente morte neuronale per
apoptosi. L’assunzione di creatina può aumentare la forza muscolare nei
pazienti con sindrome MELAS (osservati in un paziente con la sindrome MELAS
arruolati in uno studio). Effetto citotossico potenziale di
somministrazione a lungo termine.
Dicloroacetato
sodio (ceresina)
Attualmente un farmaco orfano
negli Stati Uniti. Un composto creduto per attivare il complesso della
piruvato deidrogenasi inibendo chinasi inattivante. Questo diminuisce la
produzione di lattato e promuove piruvato ossidazione. Usato per abbassare
i livelli di lattato sia nel plasma e CSF. Al momento è disponibile solo
in protocolli di ricerca. Effetto primario è quello di stimolare la
funzione di PDH inibendo chinasi che inattiva PDH. Inoltre può stimolare
glycolytic enzima fosfofruttochinasi sopprimendo inibitore allosterico
(citrato) e aumentando i livelli di attivatore (fruttosio 2,6 biphosphate) per
migliorare l’ossidazione del lattato nel fegato.
Ulteriore assistenza
ospedaliera
Ammetta di scompenso o segni di
chetoacidosi diabetica metabolica. Il diabete sembra essere la
manifestazione più comune di miopatia mitocondriale, encefalopatia, acidosi
lattica, e ictus (MELAS) sindrome. Ammetta per la gestione medica di
episodi di simil-ictus e crisi epilettiche. Ammettere di segni di aritmia
cardiaca (sindrome di Wolff-Parkinson-White), ipertensione, imminente
dissezione radice aortica, o insufficienza cardiaca congestizia (CHF) associata
a cardiomiopatia ipertrofica o dilatativa. Ammettere di segni di sindrome
nefrosica, che possono presentare in associazione con glomerulosclerosi focale
segmentale.Ammettere se un segno di addome acuto è presente; addome acuto
può essere un’indicazione di pancreatite. [13]
Ulteriori cure ambulatoriali
Monitorare attentamente
l’andamento della encefalomiopatia e sequele. Test sviluppo neurologico è
appropriato perché progressivo deterioramento intellettuale segue episodi ictus
della sindrome MELAS. Valutazione neuropsicologica è appropriato per la
presenza di disturbi dello spettro autistico (ASD).
Monitorare le curve di crescita,
perché le malattie mitocondriali, come la sindrome MELAS sono associati con
bassa statura e ritardo di crescita.
Fare riferimento al paziente di
un oftalmologo per monitorare degenerazione pigmentosa della retina, che può
essere simile a quello osservato nei pazienti con neuropatia, atassia e
sindrome retinite pigmentosa. Seguire attentamente le indicazioni (ad
esempio, oftalmoplegia, ptosi).
Monitorare attentamente le
persone con sindrome di MELAS per la perdita dell’udito con una valutazione
dell’udito, tra cui prodotti distorsione otoemissioni acustiche e del tronco
encefalico evocati uditivi risposte. Monitorare attentamente i pazienti
per cardiomiopatia e misurare Z-score per il diametro della radice aortica con
ecocardiografia. Richiedi un ECG come uno studio di riferimento per
monitorare i difetti di conduzione, anche se i pazienti sono
asintomatici. Monitorare attentamente i pazienti per il diabete di tipo 2,
ipotiroidismo, ipertiroidismo, e disfunzione paratiroidea. Monitorare
attentamente i pazienti per la persistenza di acidosi lattica.
Spettroscopia di risonanza
magnetica positroni ( 1 H-MRS) del cervello può essere
usato per monitorare potenziale efficacia terapeutica se aumentata permeabilità
della barriera emato-encefalica è una preoccupazione.
Farmaci Ospedalieri e
Ambulatoriali
Farmaci includono i seguenti:
·
Composti che possono aumentare la produzione o il trasferimento di
elettroni ATP (ad esempio, ascorbato, riboflavina, CoQ10, vitamine K-1 e K-3,
nicotinamide, creatina monoidrato)
·
Composti che possono essere utilizzati per prevenire una possibile
carenza di carnitina secondaria o disfunzione secondaria di ossidazione degli
acidi grassi (ad esempio, carnitina)
·
Composti che possono essere utilizzati per prevenire o migliorare
la progressione di episodi ictus (ad esempio, L-arginina): L-arginina potrebbe
modulare il metabolismo energetico mitocondriale inibendo glutammato
assorbimento nei mitocondri e diminuendo neurotossicità associata a disfunzione
mitocondriale ossido nitrico-mediata.
·
Composti che possono essere usati per trattare l’acidosi lattica
(ad esempio, dicloroacetato)
o
Dicloroacetato stimola la funzione piruvato deidrogenasi inibendo
piruvato chinasi deidrogenasi, l’enzima che fosforila normalmente e inattiva
piruvato deidrogenasi. Pertanto, in condizioni che provocano l’accumulo di
lattato e alanina, attivazione di piruvato deidrogenasi diminuisce il rilascio
di questi composti dai tessuti periferici e migliora il loro metabolismo
ossidativo dal fegato.
o
Questo farmaco è stato utilizzato per il trattamento di acidosi
lattica in pazienti adulti e pediatrici. Aneddotica riporta
dettagliatamente il successo del trattamento nei pazienti con sindrome MELAS.
Dicloroacetato è stato somministrato per via orale a dosi di 12,5-100 mg / kg /
d. Questo farmaco è disponibile solo in protocolli di ricerca negli Stati
Uniti.
Se sequestri si sono sviluppati
come parte della condizione, non utilizzare l’acido valproico come
anticonvulsivante, dal momento che gli episodi di pancreatite a seguito di
somministrazione valproato sono verificati e acido valproico è stato associato
a tossicità mitocondriale.
Utilizzare fenobarbital con
cautela, perché il farmaco ha dimostrato un’inibizione della catena
respiratoria in vitro.
Trasferimento
Trasferire in un centro di cura
terziario può essere richiesto di coordinare meglio la valutazione diagnostica
per includere i seguenti:
·
La biopsia muscolare
·
Valutazione per difetti enzimatici mitocondriali
·
Analisi di mutazione del mtDNA
Se la diagnosi è già nota e il
paziente è stato stabilizzato, il trasferimento può essere richiesta per una
migliore gestione delle complicanze, come la seguente:
·
Pancreatite
·
Aritmie cardiache
·
Cardiomiopatia
·
Chetoacidosi
·
Episodi di simil-ictus
Deterrenza / Prevenzione
Se le condizioni, come la
cardiomiopatia sono presenti, limitare l’esercizio. Anche se gli effetti a
lungo termine di manipolazioni dietetiche sono sconosciuti, garantire un buono
stato nutrizionale, una buona idratazione, ed evitare il digiuno come parte di
un piano di sostegno. Un grado lieve di attività aerobica può portare ad
un miglioramento della capacità aerobica. Limitare intenso esercizio
fisico a causa della possibile complicanza di rabdomiolisi.
Informazioni sulla efficacia
terapeutica dei composti riportati utilizzati come integratori nutrizionali
sono limitati; tuttavia, la maggior parte non ha effetti avversi
gravi. Gli integratori alimentari possono aiutare a prevenire un ulteriore
deterioramento in alcuni individui; Tuttavia, ulteriori ricerche è
giustificato.
Complicazioni
Le complicazioni sono i seguenti:
·
Ritardo di crescita e bassa statura
·
Progressivo deterioramento intellettuale e il declino che alla
fine può portare a demenza
·
Psicosi con la depressione, la schizofrenia, disturbo bipolare
·
Disturbi dello spettro autistico (ASD)
·
Ipoacusia neurosensoriale
·
Endocrine erettile con ipogonadismo, diabete, ipoparatiroidismo,
ipotiroidismo, ipertiroidismo e
·
CHF da cardiomiopatia e morte improvvisa da difetti di conduzione
·
Difficoltà visive legate alla degenerazione pigmentaria della
retina o cecità corticale come uno dei postumi di atrofia corticale progressiva
ed episodi di simil-ictus
·
Insufficienza renale allo stadio terminale come complicanza di
glomerulosclerosi focale segmentale
·
Insufficienza renale acuta secondaria a rabdomiolisi
·
GI disfunzione secondaria a pseudoobstruction intestinale o
pancreatite
·
Aortica dissezione root (riportato in un gruppo di affini,
richiede ulteriori studi per valutare la prevalenza)
Prognosi
Sindrome MELAS ampiamente varia
nella presentazione; tuttavia, i pazienti in generale tendono ad avere una
prognosi sfavorevole e l’esito. Il Encefalomiopatia tende ad essere grave
e progressiva demenza. Il paziente con la sindrome MELAS può finire in uno
stato di cachessia. Attualmente, esistono terapie hanno dimostrato
efficacia.
Educazione del paziente
Una volta stabilita la diagnosi,
indirizzare il paziente e la famiglia per la consulenza genetica e la
valutazione di altri membri della famiglia che possono essere a rischio di
essere colpiti.
Educare la famiglia recante
ulteriori deterioramenti e complicazioni (ad esempio, cardiomiopatia, sindrome
nefrosica, sordità, diabete, difficoltà GI) che possono influenzare i probandi
n generale, di educare la famiglia di mantenere un buono stato nutrizionale e
idratazione, e discutere le informazioni relative alle sperimentazioni in corso
(ad esempio, l’uso di dicloroacetato per persistente acidosi lattica in
pazienti con la sindrome MELAS).
- Per
eccellenti risorse di educazione del paziente, visitare il sito di
eMedicineHealthcervello e System
nervoso Centrale . Inoltre,
vedere l’articolo educazione del paziente di eMedicineHealth Stroke .
Seidowsky
A, Hoffmann M, Glowacki F, Dhaenens CM, Devaux JP, Lessore de Sainte Foy
C, et al. Renal involvement in MELAS syndrome
– a series of 5 cases and review of the literature. Clin
Nephrol. Aug 21 2012;[Medline]. - Meseguer S,
Martínez-Zamora A, García-Arumí E, Andreu AL, Armengod ME. The
ROS-sensitive microRNA-9/9* controls the expression of mitochondrial
tRNA-modifying enzymes and is involved in the molecular mechanism of MELAS
syndrome. Hum Mol Genet.
Aug 22 2014;[Medline]. - Mehrazin M, Shanske S,
Kaufmann P, Wei Y, Coku J, Engelstad K. Longitudinal changes of mtDNA
A3243G mutation load and level of functioning in MELAS. Am J Med
Genet A. Feb 15 2009;149A(4):584-7.[Medline]. [Full
Text]. - Liu K, Zhao H, Ji K,
Yan C. MERRF/MELAS overlap syndrome due to the m.3291T>C
mutation. Metab Brain Dis.
Mar 2014;29(1):139-44. [Medline]. - Testai FD, Gorelick PB.
Inherited metabolic disorders and stroke part 1: Fabry disease and
mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Arch
Neurol. Jan 2010;67(1):19-24.[Medline]. - Sasarman F, Antonicka
H, Shoubridge EA. The A3243G tRNALeu(UUR) MELAS mutation causes amino acid
misincorporation and a combined respiratory chain assembly defect
partially suppressed by overexpression of EFTu and EFG2. Hum
Mol Genet. Dec 1 2008;17(23):3697-707. [Medline]. - Nemes A, Geleijnse ML,
Sluiter W, Vydt TC, Soliman OI, van Dalen BM. Aortic distensibility
alterations in adults with m.3243A>G MELAS gene mutation. Swiss
Med Wkly. Feb 21 2009;139(7-8):117-20. [Medline]. - Scarpelli M, Zappini F,
Filosto M, Russignan A, Tonin P, Tomelleri G. Mitochondrial Sensorineural
Hearing Loss: A Retrospective Study and a Description of Cochlear
Implantation in a MELAS Patient. Genet Res Int.
2012;2012:287432. [Medline]. [Full
Text]. - [Guideline]
International Diabetes Center. Type 2 diabetes practice guidelines. 2003;[Full Text]. - Primiano G, Plantone D,
Forte F, Sauchelli D, Scaldaferri F, Gasbarrini A, et al. Acute
refractory intestinal pseudo-obstruction in MELAS: efficacy of prucalopride. Neurology.
May 27 2014;82(21):1932-4. [Medline]. - Singmaneesakulchai S,
Limotai N, Jagota P, Bhidayasiri R. Expanding spectrum of abnormal
movements in MELAS syndrome (mitochondrial encephalomyopathy, lactic
acidosis, and stroke-like episodes). Mov
Disord. Oct 2012;27(12):1495-7. [Medline]. - Fayssoil A. Heart
diseases in mitochondrial encephalomyopathy, lactic acidosis, and stroke
syndrome.Congest Heart Fail. Nov-Dec
2009;15(6):284-7. [Medline]. - Dindyal S, Mistry K,
Angamuthu N, Smith G, Hilton D, Arumugam P, et al. MELAS syndrome
presenting as an acute surgical abdomen. Ann R Coll Surg Engl.
Jan
2014;96(1):101E-103E. [Medline]. - Betts J, Jarost E,
Perry RH et al. Molecular neuropathology of MELAS; level of heteroplasmy
in individual neurons and evidence of extensive vascular involvement. Neuropathology
and Applied. Neurobiology. 2006;32:359-373. - Borner GV, Zeviani M,
Tiranti V, et al. Decreased
aminoacylation of mutant tRNAs in MELAS but not in MERRF patients. Hum
Mol Genet. Mar 1 2000;9(4):467-75. [Medline]. - Ciafaloni E, Ricci E,
Shanske S, et al. MELAS: clinical
features, biochemistry, and molecular genetics. Ann
Neurol. Apr 1992;31(4):391-8. [Medline]. - Deschauer M, Tennant S,
Rokicka A, He L, Kraya T, Turnbull DM. MELAS associated with mutations in
the POLG1 gene. Neurology.
May 15 2007;68(20):1741-2. [Medline]. - Hirano M, Pavlakis SG.
Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike
episodes (MELAS): current concepts. J Child Neurol.
Jan 1994;9(1):4-13. [Medline]. - Hirano M, Ricci E,
Koenigsberger MR, et al. Melas: an original case
and clinical criteria for diagnosis.Neuromuscul Disord. 1992;2(2):125-35. [Medline]. - Jacobs HT, Holt IJ. The
np 3243 MELAS mutation: damned if you aminoacylate, damned if you
don’t. Hum Mol Genet.
Mar 1 2000;9(4):463-5. [Medline]. - Joko T, Iwashige K,
Hashimoto T, et al. A case of mitochondrial encephalomyopathy, lactic
acidosis and stroke-like episodes associated with diabetes mellitus and hypothalamo-pituitary
dysfunction. Endocr J.
Dec 1997;44(6):805-9. [Medline]. - Kaufmann P, Engelstad
K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in MELAS: a
randomized, controlled clinical trial. Neurology.
Feb 14 2006;66(3):324-30. [Medline]. - Koga Y, Akita Y,
Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes
in MELAS.Neurology. Feb 22 2005;64(4):710-2. [Medline]. - Matsumoto J, Saver JL,
Brennan KC, Ringman JM. Mitochondrial encephalomyopathy with lactic
acidosis and stroke (MELAS). Rev Neurol Dis.
Winter 2005;2(1):30-4. [Medline]. - Pavlakis SG, Phillips
PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy,
encephalopathy, lactic acidosis, and strokelike episodes: a distinctive
clinical syndrome. Ann Neurol.
Oct 1984;16(4):481-8.[Medline]. - Pons R, Andreu AL,
Checcarelli N, Vila MR, Engelstad K, Sue CM. Mitochondrial
DNA abnormalities and autistic spectrum disorders. J
Pediatr. Jan 2004;144(1):81-5. [Medline]. - Scaglia F, Northrop JL.
The mitochondrial myopathy encephalopathy, lactic acidosis with
stroke-like episodes (MELAS) syndrome: a review of treatment
options. CNS Drugs.
2006;20(6):443-64. [Medline]. - Shanske S, Coku J, Lu
J, Ganesh J, Krishna S, Tanji K. The G13513A mutation in the ND5 gene of
mitochondrial DNA as a common cause of MELAS or Leigh syndrome: evidence
from 12 cases. Arch Neurol.
Mar 2008;65(3):368-72. [Medline]. - Shimotake T, Furukawa
T, Inoue K, Iwai N, Takeuchi Y. Familial occurrence of intestinal
obstruction in children with the syndrome of mitochondrial
encephalomyopathy, lactic acidosis, and stroke-like episodes
(MELAS). J Pediatr Surg.
Dec 1998;33(12):1837-9. [Medline]. - Sue CM, Bruno C, Andreu
AL, et al. Infantile encephalopathy associated
with the MELAS A3243G mutation. J Pediatr.
Jun 1999;134(6):696-700. [Medline]. - Tanahashi C, Nakayama
A, Yoshida M, Ito M, Mori N, Hashizume Y. MELAS with the mitochondrial DNA
3243 point mutation: a neuropathological study. Acta
Neuropathol. Jan 2000;99(1):31-8. [Medline]. - Tay SH, Nordli DR Jr,
Bonilla E, Null E, Monaco S, Hirano M. Aortic rupture in mitochondrial
encephalopathy, lactic acidosis, and stroke-like episodes. Arch
Neurol. Feb 2006;63(2):281-3. [Medline]. - Thambisetty M, Newman
NJ, Glass JD, Frankel MR. A practical approach to the diagnosis and
management of MELAS: case report and review. Neurologist.
Sep 2002;8(5):302-12. [Medline].