Osteogenesi imperfetta (s. di van der Hoeve), malattia delle ossa fragili, Malattia di Lobstein (tipo I), Sindrome di Vrolik (tipo II), Sindrome di van der Hoeve, Sindrome di Eddowes.
 |
| Michel Petrucciani (1962 – 1999)ha avuto una vita molto breve, anche a causa di una malattia congenita che lo colpì dalla nascita: osteogenesi imperfetta |
 |
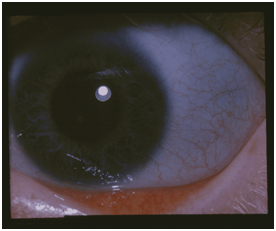 |
| Caratteristiche sclere blu nei pazienti con osteogenesi imperfetta |
 |
ln questa sindrome la sordità, di solito di tipo misto, si accompagna ai reperti di “sclere blu” e di fragilità ossea. Sono state definite una forma di tipo I, autosomica dominante, due forme (tipo Il e tipo III) autosomiche recessive, ed una forma di tipo IV con ereditarietà sia dominante che recessiva. Le principali caratteristiche istopatologiche sono una permanenza di cartilagine all’interno dell’osso encondrale, ritardo di ossificazione dello strato encondrale, e formazione ossea periosteale Anche se gli interventi sulla staffa possono ovviare alla componente trasmissiva della sordità, l’obliterazione delle finestre ovale e rotonda e l’esistenza della componente neurosensoriale, possono limitare il successo chirurgico.
L’osteogenesi imperfetta è una malattia genetica a trasmissione
autosomica dominante per anomalie nella sintesi del collagene tipo I per mutazione dei geni Col1A1 e 2. Crea problemi a carico dello scheletro, delle articolazioni, degli occhi, delle orecchie, della cute e dei denti. I fenotipi più gravi o letali sono la conseguenza di difetti genetici, che determinano molecole anomale di collagene che non riescono a formare la tripla elica.
Epidemiologia La malattia in eguali proporzioni maschi e femmine, con incidenza di 1/20-50.000 nati vivi.
Eziologia
Prevalentemente di origine genetica, tuttavia si può verificare una mutazione genetica spontanea e quindi la malattia si presenta anche con genitori sani (25% dei casi). Un genitore affetto ha il 50% di probabilità di trasmettere la malattia ai figli.
Clinica e classificazione Clinicamente manifesta fragilità ossea ed è conosciuta anche come Malattia di Lobstein (tipo I), Sindrome di Vrolik (tipo II), Sindrome di van der Hoeve, Sindrome di Eddowes.
Attualmente se ne conoscono sette tipi varianti a diversa gravità; i quattro tipi storicamente noti sono:
Tipo I: ritardo di accrescimento nel 50%, fratture ossee; cifosi e scoliosi con iperestesibilità articolare, sclere bluastre e perdita dell’udito sia a difetto neurosensoriale che a causa di anomalie ossee dell’orecchio medio e interno. In certi casi si associa a dentinogenesi imperfetta.
Tipo II: è costantemente fatale durante la vita intrauterina o nel periodo perinatale. Accentuatissima fragilità ossea con fratture multiple che si manifestano quando il feto è ancora in utero.
Tipo III: fratture alla nascita con deformazioni progressive degli arti e cifoscoliosi, sclere normali, bassa statura, dentinogenesi imperfetta comune.
Tipo IV: la forma clinicamente meno grave, con statura normale o poco ridotta, fragilità ossea lieve o moderata, fratture postnatali, sclere normali, udito normale, deformità variabili. In certi casi si associa a dentinogenesi imperfetta.
Tipo V:l’OI è una condizione autosomica dominante con una gravità simile a quello della malattia IV tipo ma una predisposizione alla formazione di callo iperplastico.
Tipi
Degli otto diversi tipi di OI, tipo I è il più comune, anche se i sintomi variano da persona a persona.
| Tipo | Descrizione | Gene | OMIM | Modalità di eredità |
| Io | mite | COL1A1 | 166.240 (IA),166200 (IB) | autosomica dominante, il 60% de novo[3] |
| II | grave e di solito letali nel periodo perinatale | COL1A1, COL1A2 | 166.210 (IIA),610.854 (IIB) | autosomica dominante, ~ 100% de novo[3] |
| III | considerato progressiva e deformante | COL1A1, COL1A2 | 259.420 | autosomica dominante, ~ 100% de novo[3] |
| IV | deformante, ma con normale sclere | COL1A1, COL1A2 | 166.220 | autosomica dominante, il 60% de novo [3] |
| V | condivide le stesse caratteristiche cliniche della IV, ma ha uniche istologici risultati (“maglia-like”) | IFITM5 | 610.967 | autosomica dominante[3][4] |
| VI | condivide le stesse caratteristiche cliniche della IV, ma ha uniche istologiche risultanze (“unghia”) | SERPINF1 | 610.968 | autosomica recessiva[3] |
| VII | associato cartilagine proteina associata | CRTAP | 610.682 | autosomica recessiva[3] |
| VIII | gravi letale, associata con la proteina leprecan | LEPRE1 | 610.915 | autosomica recessiva |
La condizione, o tipi di esso, ha avuto diversi altri nomi nel corso degli anni e in diverse nazioni. Tra alcune delle alternative più comuni sono la sindrome Ekman-Lobstein, sindrome Vrolik, e la malattia di vetro-osso colloquiale. Il nome osteogenesi imperfetta risale almeno al 1895 (K. Buday,) ed è stato il solito termine medico nel 20 ° secolo ad oggi. L’attuale sistema a quattro tipo è iniziato con Sillence nel 1979. Un vecchio sistema riteneva meno gravi i tipi “osteogenesi imperfetta Tardiva”, mentre le forme più gravi sono stati ritenuti ” osteogenesi imperfetta congenita “. Poiché questo non differenzia bene e tutti le forme sono congenite, questo da allora è caduto in disgrazia.
La condizione è stata trovata in una antica mummia egiziana del 1000 a.C.. La Norse re Ivar il senz’ossa può aver avuto questa condizione, pure. I primi studi di ‘iniziato nel 1788 con lo svedese Olof Jakob Ekman. Ha descritto la condizione nella sua tesi di dottorato e citato i casi di esso risalenti al 1678. Nel 1831, Edmund Axmann ha descritto questa patologia in se stesso e due fratelli. Jean Lobstein affrontato in adulti nel 1833. Willem Vrolik fatto un lavoro sulla malattia nel della 1850. L’idea che le forme adulte e neonatali erano la stessa è venuta nel 1897 a Martin Benno Schmidt. [24]
EPIDEMIOLOGIA
L’incidenza complessiva di OI è di circa 1 caso ogni 20.000 nati vivi; tuttavia, la forma lieve è sottodiagnosticata e la prevalenza effettiva può essere maggiore. L’incidenza sembra essere simile in tutto il mondo, anche se un tasso di aumento è stato osservato in due grandi gruppi tribali in Zimbabwe.
l’OI può presentarsi a qualsiasi età, anche se l’età in cui i sintomi (cioè, le fratture) cominciano varia ampiamente. I pazienti con forme lievi possono non avere fratture fino all’età adulta, oppure possono presentarsi con fratture nell’infanzia. I pazienti con gravi casi presentano fratture in utero.
L’OI è altrettanto comune in maschi e femmine. È stata descritta in ogni popolazione umana in cui sono stati studiati displasie scheletriche. La malattia non sembra avere alcuna predilezione per una particolare razza.
LE CAUSE:
Circa l’80-85% delle persone affette da O.I. ha una alterazione genetica (mutazione) a carico di uno dei due geni situati sui cromosomi 7 e 17, rispettivamente responsabili della produzione del collagene di tipo I. Il collagene è una proteina che può formare lunghe e resistenti fibre, che formano il tessuto connettivo del derma e delle articolazioni e costituiscono “l’impalcatura” di base su cui vengono costruite le ossa. Le mutazioni sono di vari tipi e possono comportare alterazioni della proteina collagene più o meno gravi, a seconda della loro natura molecolare e della loro posizione all’interno di uno o dell’altro dei due geni collagenici. Per questo la O.I. può presentarsi in diverse forme, a seconda del tipo di alterazione genetica. Inoltre circa 15-20% dei casi di OI non sono legati ad alterazioni della struttura del collagene direttamente ma piuttosto a mutazione a carico di geni che contribuiscono a ripiegare o a trasportare le catene di collagene.
COME SI TRASMETTE:
Per comprendere i rischi di trasmissione ereditaria dell’O.I è bene distinguere fra casi familiari (che ricorrono cioè all’interno della stessa famiglia) e casi sporadici (cioè casi isolati, che non compaiono più di una volta nella stessa famiglia). Le forme meno gravi (tipo I e IV) hanno talvolta carattere familiare e si trasmettono geneticamente secondo una modalità chiamata autosomica dominante. Questo significa che una persona affetta ogni volta che si riproduce ha il 50% di probabilità di avere un figlio affetto, indipendentemente dal sesso. Nelle malattie dominanti non esistono portatori sani: questo significa che se nessuno dei congiunti presenta segni clinici della malattia, la nascita di un figlio o di una figlia affetti da O.I è da considerarsi un evento sporadico; la nascita di figli malati da genitori sani riguardano particolarmente le forme più gravi (tipo II e III). Nei casi sporadici, l’alterazione genetica (mutazione) che causa la malattia può essere presente in forma recessiva nel patrimonio genetico dei genitori oppure non essere presente e essersi verificata solo al momento della formazione delle cellule germinali (spermatozoi o ovuli) e quindi per gli stessi genitori la possibilità di avere un altro figlio malato è variabile (25% nel primo caso, bassa e simile a quella di tutte le altre coppie sane nel secondo caso). In una piccola percentuale di casi di O.I. la probabilità di avere un altro bambino/a malato/a è però un po’ più elevata, a causa di un meccanismo genetico particolare chiamato chimerismo germinale. Quindi dato che circa il 15% dei casi di OI sono legati a forme ad ereditarietà recessiva, è sempre consigliabile che le coppie interessate si rivolgano ad un consulente genetico per poter valutare caso per caso l’eventuale rischio di ricorrenza.
LA DIAGNOSI:
La diagnosi viene posta in base a criteri clinici ed attraverso esami radiologici. Esistono test genetici sia per le forme a trasmissione autosomica dominante che per quelle ad ereditarietà recessiva ed esiste anche un test biochimico che permette di valutare le alterazioni del collagene su un piccolo pezzo di pelle prelevato al paziente, tuttavia queste indagini sono utili solo dopo la diagnosi clinica. Nel diagnosticare l’O.I., il medico dovrà escludere altre condizioni che possono presentare sintomi simili, come i deficit nutritivi, alcuni tumori, alcune malattie ereditarie (come la condrodisplasia e l’ipofosfatasia) e perfino i maltrattamenti infantili.
La diagnosi prenatale si può effettuare tramite l’ecografia, fra la 15a e la 20a settimana di gravidanza. L’esame del DNA è possibile nei casi familiari, o comunque quando sia già stata identificata l’alterazione genetica nel congiunto affetto. Generalmente soggetti OI non imparentati hanno sempre difetti genetici diversi, per questo gli aspetti clinici sono molto eterogenei.
ESISTE UNA TERAPIA:
Non esiste attualmente un trattamento risolutivo. Tuttavia, nonostante i problemi anche gravi della malattia, per le persone affette da O.I la qualità della vita può essere piena e soddisfacente, ed alcune cure possono essere di aiuto. Risultati molto incoraggianti provengono dai trattamenti, sia in età pediatrica sia in età adulta, con i bisfosfonati, farmaci che inibiscono il riassorbimento osseo, già utilizzati per la cura dell’osteoporosi degli anziani. Questo tipo di trattamento farmacologico nell’MOI ha mostrato sostanziali miglioramenti nella densità minerale ossea, nella riduzione del dolore cronico e del numero delle fratture.Per i problemi relativi a deformità scheletriche, buoni risultati vengono raggiunti con il trattamento ortopedico (tutori, corsetti, deambulatori) ed eventualmente con la terapia chirurgica. Per immobilizzare gli arti fratturati si può ricorrere ad apparecchi di trazione, invece che ai gessi, permettendo una migliore mobilità. A seconda dei casi, l’ortopedico potrà anche decidere sull’utilità di impiantare un chiodo endomidollare nei bambini più grandi, per rafforzare l’osso dall’interno. La terapia fisica e riabilitativa, sport come il nuoto, giocano un ruolo importante verso l’obiettivo di conseguire il massimo della funzionalità e della autonomia relativamente a ciascuna forma di OI.L’ipoacusia può essere trattata sostituendo lo stapedio con una protesi. Un grave problema può essere costituito dal dolore alle ossa e ai muscoli, che può deteriorare la qualità della vita, il lavoro e i rapporti sociali, e che è da combattere con tutti i mezzi a disposizione. Anche il supporto psicologico è molto importante ed aiuta le persone psicologico è molto importante ed aiuta le persone colpite da O.I. a raggiungere traguardi difficili ma non impossibili, come ad esempio quello di riuscire a mantenere una posizione eretta, almeno per un certo tempo, aiutando ad aumentare la mineralizzazione dell’osso.
RIABILITAZIONE
Il programma di riabilitazione ha lo scopo di prevenire e trattare le fratture, inducendo lo sviluppo motorio, la forza muscolare, l’ampiezza del movimento articolare, prevenendo la formazione di contratture e deformità, migliorando le abilità funzionali e l’autonomia, includendo l’uso di strumenti ortesici e correggendo I mal-allineamenti degli arti inferiori che possono interferire con il carico. L’approccio inizia con una valutazione dello sviluppo e delle capacità motorie, l’identificazione dei bisogni funzionali, la selezione degli obiettivi a breve e lungo termine e lo sviluppo di un programma individuale in linea con il raggiungimento di obiettivi specifici per ciascun paziente.
CHIRURGIA
La correzione chirurgica è un aspetto importante della gestione dei pazienti con OI. L’obiettivo è quello di migliorare la mobilità e l’autonomia. L’uso di chiodi intramidollari telescopici è lo strumento di maggior successo per la correzione delle fratture e delle deformità delle ossa lunghe. In alcuni casi possono essere necessari interventi chirurgici per la correzione di altre possibili complicanze dell’OI come le deformità della colonna e l’impronta basilare.
.
TRATTAMENTO FARMACOLOGICO
L’obiettivo della terapia è irrobustire le ossa e ridurre la loro fragilità. I pazienti devono assumere adeguate quantità di calcio e vitamina D per ottenere I migliori risultati . L’uso dei bisfosfonati endovenosi è comune. L’efficacia e sicurezza di altri approcci terapeutici sono invece ancora in discussione (orme della crescita, paratormone e bisfosfonati orali)
I bisfosfonati sono analoghi sintetici del pirofosfato, che hanno come caratteristica quella di formare un rapido e forte legame con i cristalli di idrossiapatite nella matrice minerale ossea. Essi diminuiscono il riassorbimento osseo con potenza molto diversa da un composto all’altro (Tabella II): attraverso la loro azione sugli osteoclasti, essi diminuiscono il ritmo di attivazione di nuovi processi di rimodellamento, con conseguente diminuzione delle lacune di “remodelling”.
Tra i vari bisfosfonati, il pamidronato per via endovenosa è stato studiato più approfonditamente; nella nostra esperienza il neridronato mostra analoghi effetti molto positivi. Non è ancora chiaro se i bisfosfonati per via orale siano efficaci come i preparati per via endovenosa. La terapia medica nei pazienti con OI severa va iniziata il prima possibile, anche già dall’età di un mese. Nelle forme moderate o lievi il trattamento non va eseguito in tutti i casi ma va deciso in maniera individualizzata, sulla base dell’andamento clinico, del numero di fratture, della morfologia vertebrale.
Gli effetti collaterali a breve termine della terapia con bisfosfonati si sono mostrati irrilevanti (reazione simil-influenzale dopo la prima somministrazione) mentre gli effetti a lungo termine del trattamento nei bambini sono ancora in gran parte poco conosciuti. Per questo motivo questi farmaci devono essere utilizzati solo in caso di effettiva necessità. Le modalità di trattamento ottimali, la durata, le dosi e gli intervalli di somministrazione più appropriati, il tipo di bisfosfonato, la via di somministrazione (os o ev) sono ancora oggetto di discussione.
Il trapianto di midollo osseo non ha portato a evidenze convincenti , sono tuttavia in corso studi per valutare nuovi trattamenti come gli inibitori del RANKL, un trattamento antiriassorbimento osseo reversibile, che comporta l’uso di anticorpi monoclonali contro il recettore attivatore del ligando del fattore nucleare κB; inibitori della sclerostina sono agenti anabolici che neutralizzano un inibitore della cascata del Wnt e giocano un ruolo nella differenziazione osteoblasitca senza interferire con il riassorbimento osseo.
Infine modelli murini sono ancora in studio per quanto riguarda le possibili terapie geniche (o rimpiazzando o silenziando l’allele mutato, biochimicamente trasformando delle forme severe in forme moderate).
SCHEMI DI TRATTAMENTO FARMACOLOGICO.
Per le forme I-III-IV
BAMBINI <3 aa terapia in due giorni consecutivi (giorno 1: Neridronato 1mg/kg e giorno 2 Neridronato 1mg/kg in assenza di reazione febbrile altrimenti aspettare 1-2 giorni)
BAMBINI >3 aa terapia in unica dose
OGNI 3 MESI
– Terapia con disfosfonati indicativamente Neridronato (fl da 25 o 100 mg):
- se ≤25 kg: 2 f da 25 mg in 250 ml di soluzione fisiologica, infondere 10 ml/kg lentamente in almeno 3 ore;
- se >25 kg ma <50 kg: 1 f da 100 mg in 500 di soluzione fisiologica, infondere 10 ml/kg lentamente in almeno 3 ore;
- se >50 kg: 1 f da 100 mg in 500 di soluzione fisiologica, infondere tutta la soluzione lentamente in almeno 3 ore
– Raccolta delle urine di 24 ore per ioni, citrati e creatinina, α-1-microglobulina
OGNI 6 MESI
– Esami ematochimici:
Emocromo con formula, Na, Cl, K, Ca, PO4, Azotemia, Creatinina, Sideremia, Ferritina, glicemia, Bilirubina totale e diretta, urato, colesterolo, trigliceridi, albumina, prot totali, ALT, AST, ALP, GGT, LD, ALP Ossea, IGF1, Osteocalcina, PTH, 25OH-Vitamina D, CTX, Ca-P-Creatinina-II minzione ed esame urine standard.
– Raccolta delle urine di 24 ore per Calcio, fosforo, ossalati, creatinina
– Rx colonna LL (bambini età <4 aa) o DXA con morfometria (> 4 aa)
OGNI 1-2 ANNI
– VISITA MULTIDISCIPLINARE
– VISITA ORL+AUDIOMETRIA
– VISITA ODONTOIATRICA:
– Problemi ortodontici e legati a tp con Bp:
– Conservativa
– Ecografia renale con specifica richiesta di valutare le dimensioni renali (polo-polare) e nefrocalcinosi.
La terapia verrà protratta per tre anni oltre i tre anni il follow up va valutata caso per caso.

PESS Potenziali Evocati Somestetici

PROGNOSI
Morbilità e la mortalità associate con OI variano ampiamente, a seconda del genotipo. (Vedi anche la classificazione Sillence adattato nella presentazione.) Inoltre, la variabilità avviene tra individui con differenti mutazioni, e la variabilità è stato osservato anche tra individui non imparentati con le stesse mutazioni, tra i membri della stessa famiglia, e anche tra gemelli identici in occasione .
A un estremo, si verificano i primi nati morti, e praticamente ogni osso del corpo presenta fratture multiple. La forma perinatale grave (tipo II) è di solito fatale entro poche ore dopo la nascita, anche se alcuni bambini sopravvivere per diversi mesi.All’altro estremo è OI nella sua forma più mite. In questa impostazione, gli adulti che non hanno mai sostenuto una frattura venire a cure mediche solo perché i familiari siano colpiti. Tra questi estremi è un continuum liscia di gravità.
L’aspettativa di vita dei soggetti con OI non letale sembra essere la stessa di quella per la popolazione sana, fatta eccezione per quelli con grave OI con complicazioni respiratorie o neurologiche. Anche se i pazienti con OI letale possono morire nel periodo perinatale, gli individui con estremamente grave OI possono sopravvivere fino all’età adulta.
EDUCAZIONE DEL PAZIENTE
I pazienti con OI sono generalmente ben motivati e desiderosi di raggiungere il più possibile nonostante i loro limiti fisici. L’istruzione è estremamente importante, soprattutto per quei pazienti che possono rispondere alla loro condizione in modo più negativo e quindi essere soggetto a una bassa autostima e depressione.
Educazione dei genitori e delle famiglie dei malati di OI è importante anche per aiutare ad affrontare le implicazioni giorno per giorno e la gestione continua del disturbo. Ad esempio, i genitori hanno bisogno di istruzioni speciali nella gestione di bambini colpiti. Hanno bisogno di sapere come posizionare il bambino in culla e come tenere il bambino in modo da ridurre al minimo il rischio di fratture, pur mantenendo il legame e la stimolazione fisica.
I suggerimenti riprodotti qui di seguito sono stati sviluppati dalla Fondazione Osteogenesi Imperfetta per prendersi cura dei bambini con osteogenesi imperfetta. Soprattutto, i genitori non devono sentirsi in colpa se il loro bambino si rompe un osso. I bambini devono crescere e svilupparsi, e le fratture possono verificarsi nonostante tutte le cure.
- Non abbiate paura di toccare o tenere un bambino con osteogenesi imperfetta, ma attenzione. Per sollevare un bambino con osteogenesi imperfetta, allargare le dita e mettere una mano tra le gambe e sotto le natiche, e posizionare l’altra mano dietro le spalle, collo e testa.
- Non sollevare mai un bambino con osteogenesi imperfetta da lui o lei che tiene sotto le ascelle.
- Non tirare braccia o gambe o, in quelli con grave osteogenesi imperfetta, sollevare le gambe per le caviglie a cambiare un pannolino.
- Selezionare un seggiolino per auto reclinabile. Dovrebbe essere facile da mettere o togliere il bambino nel seggiolino. Si consideri imbottitura del sedile con schiuma e utilizzando uno strato di schiuma tra il bambino e il cablaggio.
- Assicurarsi che il passeggino è grande abbastanza per ospitare calchi. Non usare un fionda o un ombrello-tipo passeggino.
- Seguire le istruzioni del medico con attenzione, soprattutto per quanto riguarda il cast di cura e di mobilità esercizi. Nuoto e
- Adulti con osteogenesi imperfetta dovrebbero evitare attività come fumare, bere, e l’assunzione di steroidi perché hanno un impatto negativo sulla densità ossea.
- Aumentare la consapevolezza di abusi sui minori e la mancanza di consapevolezza circa osteogenesi imperfetta può portare a conclusioni inesatte circa una situazione familiare. Hanno sempre una lettera del proprio medico di famiglia e una copia delle cartelle cliniche del bambino a portata di mano.
Note:
Redazione a cura di Telethon con la consulenza scientifica della prof.sa M. Mottes, Università degli Studi di Verona, Dipartimento Materno Infantile, Biologia-Genetica Policlinico “G.B. Rossi”, Piazzale L.A. Scuro, 37134, VERONA
Aggiornato e rivisto a febbraio 2003- 2008 a cura del Prof. Franco Antoniazzi Luglio 2012 della Dott.ssa Elena Monti
BIBLIOGRAFIA
- Antoniazzi F, Mottes M, Fraschini P, Brunelli PC, Tatò L. Osteogenesis Imperfecta: practical treatment guidelines. Paediatric Drugs 2000; 2:465-488.
- Devogelaer JP. New uses of bisphosphonates: osteogenesis imperfecta. Curr Opin Pharmacol 2002; 2:748-753. Bernehall Claesson I, Brodin J. What families with children with brittle bones want to tell. Child Care Health Dev 2002; 24:309-315.
- Monti E, Mottes M, Fraschini P, Brunelli PC, Forlino A, Doro F, Venturi G, Perlini S, Cavarzere P and Antoniazzi F. Current and emerging treatments for the management of osteogenesis imperfecta Ther Clin Risk Manag. 2010.;6:367-81.
Per Medici
Osteogenesi imperfetta. Antoniazzi F, Doro F, Monti E, Maines E, Morandi G. Progetto ProSE Sintesi InfoMedica via Ripamonti, 89 20141 Milano (Italy). www.progettoprose.it.
References
- Alanay Y, Avaygan H, Camacho N, Utine GE, Boduroglu K, Aktas D, et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. Apr 9 2010;86(4):551-9. [Medline]. [Full Text].
- Bargman R, Huang A, Boskey AL, Raggio C, Pleshko N. RANKL inhibition improves bone properties in a mouse model of osteogenesis imperfecta. Connect Tissue Res. Apr 2010;51(2):123-31. [Medline]. [Full Text].
- Baujat G, Lebre AS, Cormier-Daire V, Le Merrer M. [Osteogenesis imperfecta, diagnosis information (clinical and genetic classification)]. Arch Pediatr. Jun 2008;15(5):789-91. [Medline].
- Brusin JH. Osteogenesis imperfecta. Radiol Technol. Jul-Aug 2008;79(6):535-48. [Medline].
- Castillo H, Samson-Fang L. Effects of bisphosphonates in children with osteogenesis imperfecta: an AACPDM systematic review. Dev Med Child Neurol. Jan 2009;51(1):17-29. [Medline].
- Chevrel G, Schott AM, Fontanges E, et al. (2006).”Effects of oral alendronate on BMD in adult patients with osteogenesis imperfecta: a 3-year randomized placebo-controlled trial”. J. Bone Miner. Res. 21 (2): 300–6
- http://www.shriners.com/Hospitals/Chicago/conditions/OI.aspx&;usg=ALkJrhgTK_KniGQykqsPvLxmECraTW_nxg”>Chicago Shriners Hospital – Osteogenesis imperfecta” . Retrieved 2007-07-05 .
- Cole WG. The Nicholas Andry Award-1996. The molecular pathology of osteogenesis imperfecta. Clin Orthop. Oct 1997;235-48. [Medline].
- Cole WG. Advances in osteogenesis imperfecta. Clin Orthop. Aug 2002;6-16. [Medline].
- Cole WG. Bone, cartilage and fibrous tissue disorders. In: Benson MKD, Fixsen JA, MacNicol MF, Parch K, eds. Children’s Orthopaedics. 2002: 67-92.
- DiMeglio LA, Peacock M (2006). “Two-year clinical trial of oral alendronate versus intravenous pamidronate in children with osteogenesis imperfecta”. J. Bone Miner.Res. 21 (1): 132–40
- Duro Friedl EA, Ferrari Mayans L, Desalvo Portal LN, Ferrari Ruiz P, Bidondo Horno MP, Astraldi Tellechea MM. [Bruck syndrome: Osteogenesis imperfecta with congenital joint contractures.]. An Pediatr (Barc). Jul 2008;69(1):90-1. [Medline].
- http://emedicine.medscape.com/article/947588-overview&;usg=ALkJrhhd5nRW130GnEKwTOhh0M0siDCc-w”>eMedicine > Osteogenesis Imperfecta Author: Horacio Plotkin. Updated: March 2, 2010
- Esposito P, Plotkin H. Surgical treatment of osteogenesis imperfecta: current concepts. Curr Opin Pediatr. Feb 2008;20(1):52-7. [Medline].
- Forin V. [Paediatric osteogenesis imperfecta: medical and physical treatment]. Arch Pediatr. Jun 2008;15(5):792-3. [Medline].
- Francis MJ, Smith R, Bauze RJ. Instability of polymeric skin collagen in osteogenesis imperfecta. Br Med J. Mar 9 1974;1(905):421-4. [Medline].
- Fuller E, Lin V, Bell M, Bharatha A, Yeung R, Aviv RI, Symons SP. Case of the month #171: osteogenesis imperfecta of the temporal bone. Canadian Association of Radiologists Journal. 2011 November; 62(4):296-298.Gallego L, Junquera L, Pelaz A, Costilla S. Pathological mandibular fracture after simple molar extraction in a patient with osteogenesis imperfecta treated with alendronate. Med Oral Patol Oral Cir Bucal. Jun 1 2010;[Medline].
- Glorieux FH, Bishop NJ, Plotkin H, et al. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. Oct 1 1998;339(14):947-52. [Medline].
- James, William;Berger, Timothy; Elston, Dirk (2005).Andrews’ Diseases of the Skin: Clinical Dermatology .(10th ed.). Saunders. Page 517. http://en.wikipedia.org/wiki/Special:BookSources/0721629210&usg=ALkJrhjyUy-fETWpSMRgkx99WlyqSlCT2Q”>ISBN 0-7216-2921-0 Jones D, Hosalkar H, Jones S. The orthopaedic management of osteogenesis imperfecta. Clin Orthop. 2002;16:374-88.
- Gautieri A, Uzel S, Vesentini S, Redaelli A, Buehler MJ (2009). http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2718154&;usg=ALkJrhjYodPA_LBKjzfZQ17NSvSRouzRyA”>”Molecular and mesoscale disease mechanisms of Osteogenesis Imperfecta” . Biophysical Journal 97 (3): 857–865
- Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R (1998). “Cyclic administration of pamidronate in children with severe osteogenesis imperfecta”. N. Engl.J. Med. 339 (14): 947–52. http://en.wikipedia.org/wiki/Digital_object_identifier&;usg=ALkJrhjTTVx0Utd0v87gLtSBHw6ibYWE9g” title=”Digital object identifier”>doi : [Guideline] Kellogg ND. Evaluation of suspected child physical abuse. Pediatrics. Jun 2007;119(6):1232-41. [Medline]. [Full Text].
- Gregory, Ted (May 5, 2009). http://www.chicagotribune.com/health/chi-sean-stephenson-05-may05,0,4199197,full.story&usg=ALkJrhhyZMUADIZC5E-MW6jpd3_yEc5RUA”>”Osteogenesis Imperfecta: Motivational Speaker Sean Stephenson Uses His Disorder to Inspire Others” . http://en.wikipedia.org/wiki/Chicago_Tribune&;usg=ALkJrhib-X082TfoK_sifemdwSg5L2yCug” title=”Chicago Tribune”>Chicago Tribune . Retrieved August 7, 2011
- Labuda M, Morissette J, Ward LM. Osteogenesis imperfecta type VII maps to the short arm of chromosome 3. Bone. Jul 2002;31(1):19-25. [Medline].
- http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(13)61531-7/fulltext&usg=ALkJrhgqr1Nj0jGm24Pwgftle2WWbEBRfA”>Oral bisphosphonates for paediatric osteogenesis imperfecta? Lancet August 6, 2013
- “http://www.oif.org/site/PageServer%3Fpagename%3DFastFacts&;usg=ALkJrhhtmvtZ-acOdrlc_QLHBAzWFZzG7Q”>Osteogenesis Imperfecta Foundation: Fast Facts” .Retrieved 2007-07-05
- Rauch F, Travers R, Parfitt AM, Glorieux FH. Static and dynamic bone histomorphometry in children with osteogenesis imperfecta. Bone. Jun 2000;26(6):581-9. [Medline].
- Rauch F, Glorieux FH (2004) “Osteogenesis imperfecta”. Lancet 363 (9418): 1377–85. http://en.wikipedia.org/wiki/Digital_object_identifier&;usg=ALkJrhjTTVx0Utd0v87gLtSBHw6ibYWE9g” title=”Digital object identifier”>doi :http://dx.doi.org/10.1016%252FS0140-6736(04)16051-0&usg=ALkJrhg_lEz93lAXaQCCqmumY3dFY1v0KQ”>10.1016/S0140-6736(04)16051-0 . http://en.wikipedia.org/wiki/PubMed_Identifier&;usg=ALkJrhiTaTJodZBVwy6rDgv6hqsxcB73Vg” title=”PubMed Identifier”>PMID http://www.ncbi.nlm.nih.gov/pubmed/15110498&;usg=ALkJrhjAsxTEMD1R2L4y209re-c8q3We0w”>15110498 Rauch F, Munns C, Land C, Glorieux FH. Pamidronate in children and adolescents with osteogenesis imperfecta: effect of treatment discontinuation. J Clin Endocrinol Metab. Apr 2006;91(4):1268-74. [Medline].
- http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(13)61091-0/abstract&usg=ALkJrhipqGQwG2hIfcUG9hnCdiG65IkbmQ”>Risedronate in children with osteogenesis imperfecta: a randomised, double-blind, placebo-controlled trial , Lancet, 6 August 2013
- Salehpour S, Tavakkoli S. Cyclic pamidronate therapy in children with osteogenesis imperfecta. J Pediatr Endocrinol Metab. Jan-Feb 2010;23(1-2):73-80. [Medline].
- Shapiro JR, Thompson CB, Wu Y, Nunes M, Gillen C. Bone Mineral Density and Fracture Rate in Response to Intravenous and Oral Bisphosphonates in Adult Osteogenesis Imperfecta. Calcif Tissue Int. Jun 11 2010;[Medline].
- Shapiro JR, Lietman C, Grover M, Lu JT, Nagamani SC, Dawson BC, Baldridge DM, Bainbridge MN, Cohn DH, Blazo M, Roberts TT, Brennen FS, Wu Y, Gibbs RA, Melvin P, Campeau PM, Lee BH (2013) Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation. J Bone Miner Res doi: 10.1002/jbmr.1891
- Sillence D. Osteogenesis imperfecta: an expanding panorama of variants. Clin Orthop. Sep 1981;11-25.[Medline].
- Sillence DO, Senn A, Danks DM. Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. Apr 1979;16(2):101-16. [Medline].
- Smith R, Francis MJ, Houghton GR. The brittle bone syndrome. In: Osteogenesis Imperfecta. London: Butterworth. 1983.
- Sofield HA, Page MA, Mead NC. Multiple osteotomies and metal-rod fixation for osteogenesis imperfecta.J Bone Joint Surg. 1952;34A:500-2.
- Steiner, RD; Pepin, MG, Byers, PH, Pagon, RA, Bird, TD, Dolan, CR, Stephens, K, Adam, MP (January 28, 2005). http://www.ncbi.nlm.nih.gov/books/NBK1295/&;usg=ALkJrhge22pFUkhUzkqDnWkEabdRg55yDw”>Osteogenesis Imperfecta . http://en.wikipedia.org/wiki/PubMed_Identifier&;usg=ALkJrhiTaTJodZBVwy6rDgv6hqsxcB73Vg” title=”PubMed Identifier”>PMIDhttp://www.ncbi.nlm.nih.gov/pubmed/20301472&;usg=ALkJrhjKZrNYFuAHf3iVblJlofYeq-F1Ew”>20301472 . Retrieved 26 March 2012 .
- Viljoen D, Beighton P (1987). “Osteogenesis imperfecta type III: an ancient mutation in Africa?”. Am. J. Med.Genet. 27 (4): 907–12.
- Ward LM, Rauch F, Travers R. Osteogenesis imperfecta type VII: an autosomal recessive form of brittle bone disease. Bone. Jul 2002;31(1):12-8. [Medline].
- Wekre LL, Frøslie KF, Haugen L, Falch JA. A population-based study of demographical variables and ability to perform activities of daily living in adults with osteogenesis imperfecta. Disabil Rehabil. 2010;32(7):579-87. [Medline].
- Zeitlin L, Fassier F, Glorieux FH. Modern approach to children with osteogenesis imperfecta. J Pediatr Orthop B. Mar 2003;12(2):77-87. [Medline].