sindrome di Norrie NDP,Sindrome di Norrie-Warburg , pseudoglioma , Atrofia Bulborum ereditaria , Episkopi cecità
Che cosa è la malattia Norrie?
|
|
|
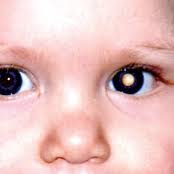
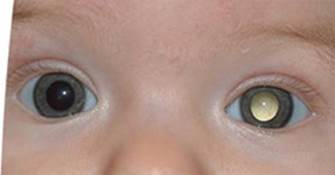
La malattia o sindrome di Norrie è una malattia genetica molto rara, legata al gene recessivo NDP presente sul cromosoma X, che causa cecità, sordità e ritardo mentale. Colpisce solo i maschi, mentre le femmine sono portatrici sane. Colpisce principalmente l’ occhio (displasia vitreo-retinica) e porta quasi sempre alla cecità, nei neonati maschi alla nascita o subito dopo la nascita. colpisce solo i maschi, neonati presso o subito dopo la nascita, perché la malattia è ereditaria recessiva X-linked mentre le femmine portatrici hanno un fenotipo normale. Sin dalle prime settimane di vita si manifesta leucocoria bilaterale, [Leucocoria (anche leucocoria [1] o riflesso pupillare bianco ) è un riflesso bianco anomalo dalla retina dell’occhio], dovuta alla massa bianco-giallastra della retina immatura in posizione retro-cristallinica, con presenza di pochi vasi e processi ciliari allungati sulla sua superficie. Occasionalmente la camera anteriore è stretta. La malattia si associa a microftalmia, ipoplasia dell’iride, sinechie, glaucoma e cataratta. In un terzo dei casi, verso i 20-30 anni, si sviluppa sordità percettiva bilaterale. A causa della rapida evoluzione verso l’atrofia del globo (tisi) nell’arco di qualche mese, non può essere attuata una cura e, di conseguenza, la cecità è precoce. È comune il ritardo psicomotorio (65%) e si possono associare varie anomalie sistemiche (cardiache, polmonari, scheletriche, uro-genitali e gastrointestinali). La diagnosi prenatale è affidabile, in quanto identifica le femmine portatrici a rischio. Il gene-malattia è stato recentemente scoperto e mappa sul cromosoma Xp11.4. La consulenza genetica ha un ruolo determinante.
QUANTO È DIFFUSA LA MALATTIA DI NORRIE?
Norrie malattia è una malattia rara; la sua incidenza esatta non è nota. Non è associata ad alcun gruppo razziale o etnico specifico.
SINTOMI
I sintomi più importanti della malattia Norrie sono oculare.
Alla nascita o subito dopo, nei primi mesi di vita, si nota in entrambi gli occhi una massa bianco-giallastra, pseudo-tumorale, costituita da residui di vasi sanguigni e tessuto retinico imperfetto, che “bloccano” la luce in entrata. Questo materiale, che comprende già forse un distacco della retina , può essere confuso con un tumore, e quindi viene chiamato pseudoglioma . [ 1 ] [ 4 ] Il primo sintomo tipicamente visibile è un riflesso pupillare bianco (leukokoria; pseudoglioma). La malattia ha una forte variabilità (anche all’interno di una stessa famiglia) e ulteriori manifestazioni oculari possono includere periferia retinica avascolare, emorragie vitreoretiniche, pieghe retiniche, distacco retinico essudativo e/o trazionale e persistente vascolarizzazione fetale.
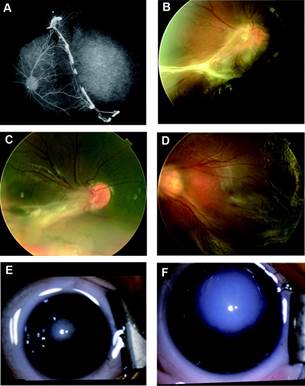
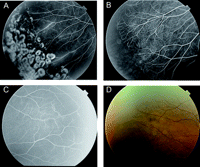
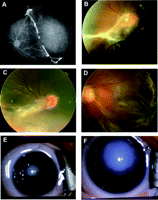
Fotografie fondo e angiografia con fluoresceina dei pazienti.(A) fluoresceina angiogramma dell’occhio sinistro della probanda della famiglia 1 (III-1), che mostra neovascolarizzazione e una grave avascularization retinica tipica di FEVR. Immagine (B)Fundus del diritto occhio del probando della famiglia 2 (III-1), che mostra una macula trascinato con persistente residuo hyaloid. (C) occhio destro della probanda della famiglia 3 (III-3) mostra una ricerca simile a quello mostrato in (B). Immagine (D) fondo dell’occhio sinistro della probanda della famiglia 4 (III-1) mostra un distacco di retina totale. (E)fotografia esterna l’occhio destro di probando della famiglia 5 (III-1) che mostra fibroplasia retrolental. (F) fotografia esterno dell’occhio sinistro della probanda della famiglia 6 (III-1) che mostra una camera anteriore piatta con opacità corneale progressione di buphthalmia.
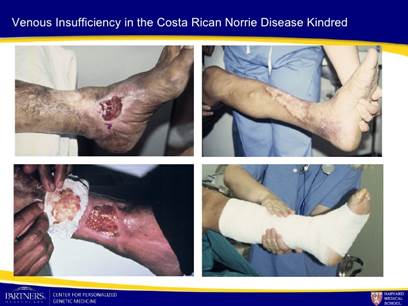
La cornea, l’iride, il tessuto ciliato e l’epitelio pigmentato retinico possono essere danneggiati dalla malattia. La malattia si presenta anche con un maggior rischio di anomalie del sistema periferico vascolare (es: insufficienza venosa).
Il primo dato visibile è la leucocoria , un riflesso pupillare grigio-giallo che nasce da una massa di tessuto non organizzata dietro la cornea .Tale massa viene spesso accresciuta a causa di sanguinamenti che lasciano residui cicatriziali nel fondo oculare e che sono causa nella quasi totalità dei casi di un distacco retinico di tipo emorragico. Non vi sono cure o rimedi chirurgici per la malattia, in quanto l’occhio è molto malformato, e tende negli anni a peggiorare. Solo nei casi in cui il distacco retinico non sia totale si può tentare di intervenire per preservare almeno la percezione della luce. La malattia ha carattere degenerativo e con l’età insorgono cataratta, degenerazione della cornea, sanguinamento, atrofia dell’iride, adesioni, nistagmo, atrofia del bulbo oculare e glaucoma. L’aspetto dell’occhio nella fase finale della malattia, verso gli 8-10 anni è spesso di un occhio piccolo (microftalmia), molto chiaro, con una pupilla di ridotte dimensioni. Intervenire chirurgicamente o con trattamento laser può essere necessario per la riduzione della pressione oculare qualora il paziente presenti glaucoma; spesso i pazienti sono però semplicemente sottoposti a cure con colliri specifici per la riduzione della pressione oculare. Nell’ultima fase della malattia Norrie, i globi appaiono piccoli e infossati in ( tisi bulbare ) e la cornea sembra essere lattiginosa. [ 1 ]
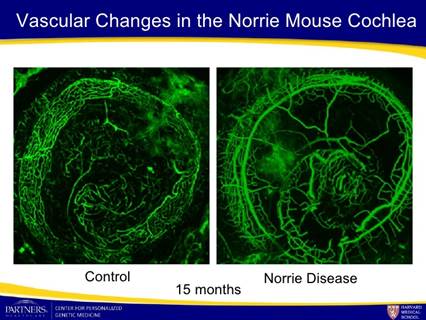
La maggior parte delle persone affette sviluppa negli anni anche una forma di sordità precoce è neurosensoriale , mite e asimmetrica. [ 1 ], legata alla degenerazione progressiva dei vasi sanguigni che irrorano la coclea.
|
|
|
Titolo: R76 Norrie Disease Capitolo: difetti di sviluppo sezione Capitolo: sindromica Perdita dell’udito / di Norrie Disease TB Caso numero: 532 Sesso: Maschio (. anni) Età: 77 otologico Dx:1. Sindrome di Norrie, caratterizzata da perdita uditiva profonda, bilaterali 2. Grave degenerazione dell’organo del Corti, membrana tettoria, stria vascularis, e neuroni cocleari, orecchio destro cisti aracnoidea del condotto uditivo interno |
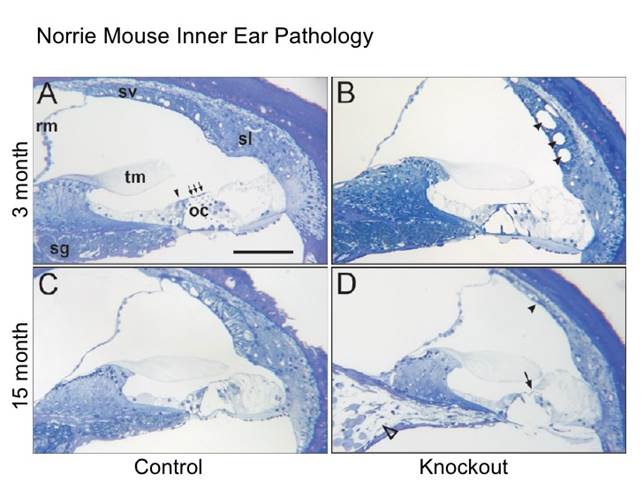
Dati audiologici indicano che la degenerazione dell’orecchio è probabilmente influenzata dalla degenerazione delle cellule cigliate, o recettori sensoriali, della coclea. Sono in genere efficaci gli ausili all’udito normalmente adottati, che stimolano i neuroni ancora attivi, o l’impianto cocleare nei casi in cui non sia più di aiuto la normale tecnologia protesica. L’insorgenza della sordità può essere molto variabile, dall’infanzia, all’adolescenza, sino ai 20-30 anni. Una volta iniziato il processo degenerativo si raggiungerebbe dapprima una perdita delle frequenze “alte”, con una perdita lieve e asimmetrica, fino ad avere una sordità profonda, bilaterale, simmetrica verso i 35-40 anni. Alcuni pazienti lamentano anche periodi di perdita improvvisa dell’udito, bi o mono laterale, che può durare da qualche ora a qualche giorno, di incerta eziologia, che normalmente si risolvono spontaneamente o con l’aiuto di cortisonici. Caratteristica peculiare è comunque una buona capacità di riconoscere suoni e parole anche quando la perdita dell’udito è profonda. Non è di particolare utilità l’apprendimento del linguaggio dei segni.
per una dettagliata descrizione delle caratteristiche uditive della malattia si può fare riferimento allo studio di Sims K, Rehm H (2005) et al. “Audiologic features of Norrie disease” [www.ncbi.nlm.nih.gov/pubmed/16134349]
Circa il 30-50% delle persone affette dalla malattia presentano ritardo psicomotorio, anomalie comportamentali, psicosi, comportamenti “simil autistici”, ritardo mentale, a volte di tipo regressivo dopo una prima fase di sviluppo normale. Non è ancora completamente chiaro se ciò sia il risultato diretto della mutazione genetica o essa sia solo una concausa, e che invece concorrano fattori “ambientali”, quali la cecità, il senso di isolamento percettivo, la sordità, la difficoltà di relazione con la madre e la famiglia che subisce un trauma alla nascita di un figlio con minorazione visiva, altri traumi quali le continue ospedalizzazioni o un insufficiente intervento educativo specifico. I principali studi a supporto della teoria regressiva sono quelli svolti a cavallo degli anni 60-70 da M. Warburg, che hanno però il limite di essere stati effettuati su pazienti per lo più istituzionalizzati precocemente, e sottoposti quindi ad un trauma di separazione familiare.
Vi sono almeno 70 differenti mutazioni genetiche certificate, ma non è ancora stato riscontrato un rapporto diretto tra la specifica mutazione e la possibilità di insorgenza del ritardo mentale o della gravità del problema uditivo. Studi evidenziano tuttavia come vi sia grande variabilità anche tra familiari portatori della stessa mutazione. Circa il 15% dei pazienti presenta una delezione del gene o di una parte del gene, invece di una mutazione, e in tali casi si ha più frequentemente ritardo mentale grave, epilessia, mioclono, crescita somatica ridotta, pubertà tardiva, problematiche di sviluppo sessuale, microcefalia. [1] Tali delezioni possono coinvolgere anche i vicini geni MAO-A MAO-B e risultare in un quadro neurologico complesso.
In rari casi in tarda età si possono sviluppare problematiche venose-arteriose come varici, problemi alla circolazione periferica. La salute generale non è intaccata dalla malattia, che non ha alcuna incidenza sul sistema immunitario del paziente. Il range della vita è assolutamente normale.
È possibile certificare la patologia tramite un test genetico, ed è possibile anche la diagnosi pre-impianto. In Italia, in centri specializzati è possibile la ricerca dell’anomalia genetica anche sull’ovocita, senza intervenire sull’embrione.
Le persone malate devono sottoporsi con frequenza a controlli oculistici e audiometrici, in modo da intervenire a contrastare immediatamente ogni eventuale problematica aggiuntiva. Non è ancora stata identificata una precisa cura, ma sono in corso ricerche, tra cui una in Italia sotto l’egida della Fondazione Telethon, per meglio comprendere i processi che coinvolgono il paziente esistono anche studi genetici in corso negli Stati Uniti, ad esempio quello di Ohlmann et al (2005), o quello legato al prof. N.Jeremy [2]
Di grande importanza un intervento educativo che miri ad infondere nel bambino autostima, fiducia, capacità di comunicazione e movimento, sviluppo multisensoriale e che sia di supporto alle famiglia aiutandole nel processo educativo. Esistono associazioni di supporto per gli ammalati e le loro famiglie[1]. A supporto dei pazienti si è creato qualche anno fa un forum a cui aderiscono più di un centinaio di iscritti, tra pazienti e famiglie di bambini affetti [3]. Esso ha lo scopo di informare, di condividere studi, idee, consigli pratici e emozioni di quanti sono coinvolti dalle problematiche legate alla Sindrome. Come riportato anche ufficialmente dalla Norrie Disease Association le persone affette da sindrome di Norrie possono richiedre spesso ausilio e attenzione esterne, ma sono spesso in grado di vivere una vita autonoma e soddisfacente grazie ad un intervento precoce e a moderni mezzi tecnologici.
Come fanno le persone ad ereditare la malattia di Norrie?
|
|
|
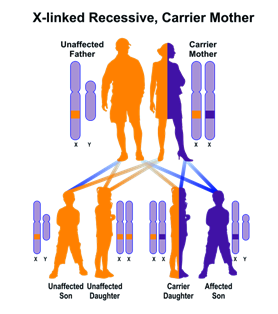
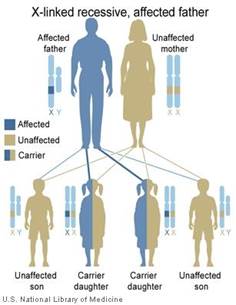
Figura 1: L‘ereditarietà legata al cromosoma X. (A) Madre portatrice. (B)
Padre colpite. Immagini modificate dalla National Library of Medicine degli
Stati Uniti. (http://ghr.nlm.nih.gov/handbook/illustrations).
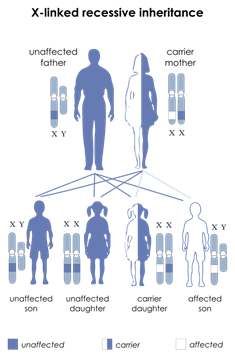
Ereditarietà recessiva X-linked
Norrie malattia è una malattia genetica causata da mutazioni nel NDP gene, che si trova su Xp11.4 (GeneID: 4693). [ 5 ] Si è ereditata come recessiva X-linked modo. Ad ogni gravidanza di una donna portatrice sana si ha il 50% delle probabilità che un figlio maschio sia malato, e 50% di probabilità che sia sano, avendo ereditato il cromosoma X non affetto. Una figlia femmina avrà il 50% di probabilità di essere sana e il 50% di probabilità di essere portatrice sana, a seconda di quale cromosoma X abbiano ereditato dalla madre. I figli di un maschio con la malattia saranno tutti sani, ereditando il cromosoma X dalla madre, mentre le figlie saranno tutte portatrici sane.Ciò significa che quasi solo i maschi sono colpiti. Figli di uomini colpiti non hanno la mutazione, mentre tutte le loro figlie saranno portatori genetici della mutazione. Inoltre di solito non mostrano sintomi clinici, ma erediteranno la mutazione al 50% della loro prole. Figlie che ricevono il gene mutato sarà anche, come la madre, i portatori asintomatici, ma il 50% dei loro figli esprimeranno sintomi clinici.
Le femmine sono molto improbabile per esprimere i segni clinici. Un possibile scenario che porta a questo (improbabile) caso sarebbe se entrambe le loro copie dei NDP mutazioni gene orso, che potrebbe essere il caso in consanguinee famiglie o per una spontaneamutazione somatica . Un’altra spiegazione per le femmine colpite potrebbe essere distortainattivazione del cromosoma X .
Si contano circa 200 casi noti al mondo, anche se c’è chi ipotizza ci siano 5-600 casi[senza fonte]. Vi è tuttavia la possibilità che nasca un individuo affetto da modificazione del gene NDP anche in assenza di una storia familiare di Malattia di Norrie, a seguito di mutazioni spontanee del gene, chiamate “de novo mutations”. Tali persone sono comunque portatrici del gene mutato al pari di chi lo ha ereditato. (www.geneclinics.org/profiles/norrie/details.html).
Tuttavia, ci sono stati alcuni rari casi in cui le donne hanno mostrato sintomi associati alla malattia di Norrie, come anomalie retiniche e la perdita dell’udito lieve. [ 4 ]
GENETICA QUALI GENI SONO LEGATI ALLA MALATTIA NORRIE?
IL GENE NDP
Una mutazione nel NDP gene causa la malattia Norrie. Il nome ufficiale del gene è “malattia Norrie (pseudoglioma)”, e il suo simbolo ufficiale è NDP . La funzione normale del NDP gene è quello di produrre le istruzioni per la creazione di una proteina chiamata Norrina . La Norrina partecipa nella cascata Wnt, una sequenza di passi che influenzano il modo in cui le cellule ed i tessuti si sviluppano che è importante per la divisione cellulare (proliferazione), attacco di cellule tra loro (adesione), movimento cellulare (migrazione), e molte altre attività cellulari.
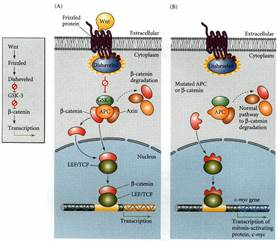
Trasduzione del segnale Wnt. (A) La proteina Wnt si lega al suo recettore, un membro della famiglia di proteine Frizzled. La proteina Frizzled quindi attiva Disheveled, consentendole di diventare un inibitore di glicogeno sintasi chinasi 3 (GSK-3). GSK-3, se fosse attiva, impedirebbe la dissociazione di β-catenina dalla proteina APC. Così, inibendo GSK-3, il segnale Wnt libera β-catenina da associare a una proteina LEF per diventare un fattore di trascrizione attiva. (B) In cellule adulte, se il gene per APC o β-catenina è mutato tale che non possono legare insieme, β-catenina è costitutivamente consentito nel nucleo. Questo fa sì che per attivare alcuni geni divisione cellulare e avviare i tumori. (B dopo Pennisi 1998 .)
 La Norrina è una delle molte proteine, o ligandi, che si può attaccare (bind) ad altre proteine chiamate recettori frizzled. Questi recettori sono incorporati nelle membrane esterne delle cellule. La Norrina si lega con il recettore frizzled-4 (prodotto dal FZD4 gene), montaggio insieme come una chiave in una serratura. Quando un ligando si lega ad un recettore frizzled, avvia un processo multi-step che regola l’attività di alcuni geni. In particolare, sembra che la Norrina svolga un ruolo critico nella specializzazione delle cellule della retina per le loro capacità sensoriali uniche. Al fine di avviare la cascata di Wnt, la Norrina deve associarsi (collegare) ad un’altra proteina chiamata frizzled-4. Le mutazioni nella proteina Norrina interferire con la sua capacità di legarsi ai frizzled-4, con La proteina Norrina e del suo recettore frizzled-4 partecipare ai processi di sviluppo che si ritiene siano cruciali per il normale sviluppo degli occhi e altri sistemi del corpo. In particolare, sembra Norrina a svolgere un ruolo critico nella specializzazione delle cellule della retina (lo strato sottile nella parte posteriore dell’occhio che rileva la luce e colore) e l’istituzione di un afflusso di sangue alla retina e l’orecchio interno.. E ‘anche coinvolto nella creazione di un apporto di sangue ai tessuti della retina e l’orecchio interno, e lo sviluppo di altri sistemi del corpo. Essa è essenziale nello sviluppo dei vasi sanguigni che alimentano le cellule della retina e ne consentono lo sviluppo in fase intrauterina. [ 6 ] Legandosi ad altre proteine fa sì che prima della nascita nel feto regrediscano quei vasi sanguigni che avvolgono le strutture dell’occhio, e dunque, nei casi in cui manca tale proteina, l’occhio si sviluppa in modo imperfetto: la retina risulta fortemente malformata, e spesso il corpo vitreo è “primitivo”. Inoltre il suo ruolo appare importante nel corretto funzionamento dei vasi sanguigni che compongono la stria vascularis, ovvero quell’insieme di vasi sanguigni presenti nell’orecchio interno. Non è invece chiaro quale ruolo abbia nello sviluppo del cervello.
La Norrina è una delle molte proteine, o ligandi, che si può attaccare (bind) ad altre proteine chiamate recettori frizzled. Questi recettori sono incorporati nelle membrane esterne delle cellule. La Norrina si lega con il recettore frizzled-4 (prodotto dal FZD4 gene), montaggio insieme come una chiave in una serratura. Quando un ligando si lega ad un recettore frizzled, avvia un processo multi-step che regola l’attività di alcuni geni. In particolare, sembra che la Norrina svolga un ruolo critico nella specializzazione delle cellule della retina per le loro capacità sensoriali uniche. Al fine di avviare la cascata di Wnt, la Norrina deve associarsi (collegare) ad un’altra proteina chiamata frizzled-4. Le mutazioni nella proteina Norrina interferire con la sua capacità di legarsi ai frizzled-4, con La proteina Norrina e del suo recettore frizzled-4 partecipare ai processi di sviluppo che si ritiene siano cruciali per il normale sviluppo degli occhi e altri sistemi del corpo. In particolare, sembra Norrina a svolgere un ruolo critico nella specializzazione delle cellule della retina (lo strato sottile nella parte posteriore dell’occhio che rileva la luce e colore) e l’istituzione di un afflusso di sangue alla retina e l’orecchio interno.. E ‘anche coinvolto nella creazione di un apporto di sangue ai tessuti della retina e l’orecchio interno, e lo sviluppo di altri sistemi del corpo. Essa è essenziale nello sviluppo dei vasi sanguigni che alimentano le cellule della retina e ne consentono lo sviluppo in fase intrauterina. [ 6 ] Legandosi ad altre proteine fa sì che prima della nascita nel feto regrediscano quei vasi sanguigni che avvolgono le strutture dell’occhio, e dunque, nei casi in cui manca tale proteina, l’occhio si sviluppa in modo imperfetto: la retina risulta fortemente malformata, e spesso il corpo vitreo è “primitivo”. Inoltre il suo ruolo appare importante nel corretto funzionamento dei vasi sanguigni che compongono la stria vascularis, ovvero quell’insieme di vasi sanguigni presenti nell’orecchio interno. Non è invece chiaro quale ruolo abbia nello sviluppo del cervello.
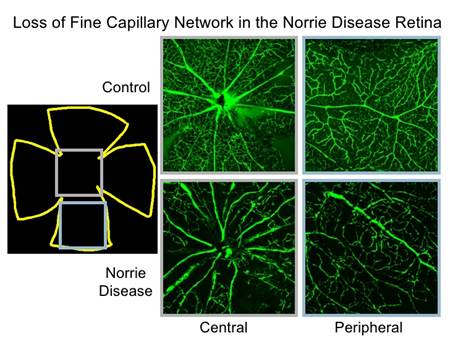
Immagini fondo e angiografia con fluoresceina degli occhi dei portatori femminili in famiglia 3. fluoresceina angiografia della madre (A) e la sorella III-4 (B)che mostra i segni tipici di FEVR, come mancanza di vascolarizzatine.e tortuosità dei vasi retinici. Le altre due sorelle III-1 (C) e III-2(D) hanno mostrato minimo tortuosità vaso retinico senza vascolarizzatine.
La Norrina non è solo importante nello sviluppo dell’occhio. La mutazione del NDP gene può influenzare gli altri sistemi del corpo. I problemi più gravi sono causati da delezioni cromosomiche nella regione del NDP gene, causando la prevenzione del prodotto del gene, o anche che i vicini MAO geni. Quando le mutazioni cambiano semplicemente un singolo aminoacido in Norrin, gli effetti sono meno diffuse e gravi. Tuttavia, la posizione e il tipo di NDP mutazione non significa necessariamente determinano il grado di gravità della malattia, in quanto altamente variabili segni clinici sono stati diagnosticati in pazienti portatori la stessa mutazione. Pertanto, il coinvolgimento di altri geni modificatori è molto probabile. D’altra parte, se certe strutturalmente importanti amminoacidi vengono modificati (ad esempio le cisteine che formano il putativo cistina nodo ), il risultato clinico ha dimostrato di essere più gravi. [ 7 ]
COME I CAMBIAMENTI NEL GENE NDP CORRELANO ALLE MALATTIE DEGLI OCCHI?
familiare vitreoretinopatia essudativa – causata da mutazioni nel gene NDP
Diversi mutazioni del gene NDP sono stati trovati come causa della malattia degli occhi: vitreoretinopatia essudativa familiare. Queste mutazioni cambiano mattoni delle proteine singole (aminoacidi) nella proteina Norrina, alterando la normale piegatura di Norrina o impedendole di legarsi ai frizzled-4. Il Norrina difettosa interrompe la segnalazione chimica nello sviluppo dell’occhio, che interferisce con la formazione di vasi sanguigni ai bordi della retina. Il conseguente apporto di sangue anormale di questo tessuto porta a danni alla retina e la perdita della vista in alcune persone con la vitreoretinopatia familiare essudativa.
Sindrome di Norrie – causata da mutazioni nel gene NDP
Più di 75 mutazioni nel NDP gene sono state identificate nelle persone con malattia Norrie. Queste mutazioni influiscono sulla capacità della proteina Norrin di legarsi con frizzled-4, interferendo con la specializzazione delle cellule retiniche per la loro funzione sensoriale unica. Come risultato, masse di cellule retiniche immature si accumulano nella parte posteriore degli occhi. Turbativa del ruolo di Norrin nella creazione di vasi sanguigni che irrorano il occhio provoca infine alcuni dei tessuti per abbattere.
La Norrina si esprime anche in altri sistemi del corpo, e gli effetti della malattia possono essere diffusa, tra cui ritardo mentale, crisi epilettiche, problemi comportamentali, e ritardo dello sviluppo. Anomalie specifici e la loro gravità dipendono dal tipo e la posizione del NDP mutazione del gene. Le mutazioni che cancellano parti del NDP gene impediscono la produzione di Norrin e provocano gravi problemi che interessano molti sistemi del corpo, oltre agli occhi. Mutazioni che eliminare o modificare singoli aminoacidi solito risultato in effetti meno diffusi.
Altre distrofie retiniche – causata da mutazioni nel gene NDP
NDP mutazioni genetiche possono causare altri disturbi che colpiscono la retina. Una mutazione è associata a un disturbo chiamato malattia Coats. Questo disturbo provoca perdita di vasi sanguigni nella retina e distacco della retina, una condizione in cui strati della retina separata, con conseguente perdita della vista. Persistente del vitreo iperplastico primitivo (PHPV) è un altro disturbo della retina che può essere causata da NDP mutazioni geniche. In persistente del vitreo iperplastico primitivo, un residuo di un vaso sanguigno trovato nell’occhio prima della nascita rimane come un gambo bianco fibrosa tra la parte posteriore dell’occhio e la lente. Persistente del vitreo iperplastico primitivo può causare la perdita della vista attraverso il distacco della retina, nuvolosità del cristallino (cataratta) o un aumento della pressione all’interno dell’occhio (glaucoma), che può danneggiare il nervo ottico.
Inoltre, NDP mutazioni geniche possono influenzare il corso di una malattia retinica che colpisce alcuni neonati prematuri. Retinopatia del prematuro è una condizione in cui i vasi sanguigni anomali vengono visualizzati nella retina e possono causare il distacco di retina. I bambini con retinopatia della prematurità possono sperimentare un miglioramento delle condizioni nel corso del tempo, ma alcuni NDP mutazioni del gene sono state associate con un peggioramento della condizione.
Dove si trova il NDP trova gene?
Citogenetica Località: Xp11.4
Location molecolare sul cromosoma X: coppie di basi 43.948.775 a 43.973.674
![]()

Il NDP gene è situato sul breve (p) braccio cromosoma X nella posizione 11.4.
Più precisamente, l’ NDP gene si trova dalla coppia di basi 43.948.775 a 43.973.674 coppia di basi sul cromosoma X.
DIAGNOSI
Norrie malattia e altre malattie legate NDP sono diagnosticati con la combinazione dei risultati clinici e test di genetica molecolare.Test genetici molecolari identifica le mutazioni che causano la malattia in circa il 85% dei maschi affetti. [ 1 ] Le diagnosi cliniche si basano su oculari risultati. Norrie malattia viene diagnosticata quando masse fibrovascolari grigio-giallo si trovano dietro l’occhio dalla nascita fino a tre mesi. I medici cercano anche per la progressione della malattia da tre mesi con 8-10 anni di età. Alcune di queste progressioni comprendono cataratta, iris atrofia , shallowing della camera anteriore , e contrazione del globo. [ 1 ] A questo punto, le persone con la condizione o hanno solo la percezione di luce o nessuna visione a tutti.
Analisi genetiche molecolari viene utilizzato per più di una diagnosi iniziale. Viene utilizzato per confermare i test diagnostici , per le femmine di prova del vettore, prenatale diagnosi e diagnosi genetica preimpianto. Ci sono tre tipi di test di genetica molecolare clinica. In circa l’85% dei maschi, Mis-sense e splicing mutazioni del gene NDP e delezioni geniche parziali o interi vengono rilevati utilizzando analisi di sequenza. [ 1 ] Analisi cancellazione / duplicazione può essere utilizzato per rilevare il 15% delle mutazioni che sono submicroscopici delezioni . Questo è anche utilizzato durante il test per portatore femmine. L’ultimo test utilizzato è linkage analisi , che viene usato quando le prime due sono disponibili. Le analisi di linkage è consigliato anche per quelle famiglie che hanno più di un membro colpito dalla malattia. [ 1 ]
Test Genetico
Un test genetico può confermare la diagnosi, ed è disponibile per valutare il rischio genetico per i membri della famiglia nella consulenza prenatale. Il gene di Norrie, l’ N D P, è formato da tre esoni e codifica una proteina composta da 133 amminoacidi. Attraverso il sequenziamento diretto del DNA, è possibile individuare nella maggior parte dei soggetti maschili le mutazioni nel gene N D P che possono causare la
malattia. Nei casi in cui l’esistenza della malattia di Norrie non possa essere confermata dall’analisi del DNA, è opportuno compiere un’indagine su altri geniche sono stati associati con condizioni cliniche simili(FZ D 4 , L R P 5 , T S P A N 1 2 ).
Diagnosi Differenziale
Diverse altre malattie possono essere facilmente confuse con la Malattia di Norrie, tra le quali il retinoblastoma, la displasia retinica infantile, il vitreo iperplastico primitivo, la retinopatia del prematuro, la displasia retinica di Reese, la sindrome di Coats, la
retinoschisi giovanile legata al cromosoma X, la sindrome osteoporosi-pseudoglioma, e specialmente la vitreoretinopatia essudativa familiare (FEVR). Quest’ultima malattia è stata associata alla mutazione di quattro geni diversi (NDP, FZD4, LRP5, TSPAN 12). Per questo motivo il test genetico è fondamentale per una corretta diagnosi.
Terapia
Nella maggior parte dei casi, nel momento in cui la malattia viene notata si è già verificato un distacco
retinico totale e irreversibile. Tuttavia, i pazienti che non hanno perso completamente la vista possono essere operati chirurgicamente o con laser, anche in tenera età .La perdita uditiva invece può essere trattata con ausili acustici o con impianti cocleari. Consulenza psicologica, terapia farmacologica o comportamentale e una cura da parte di educatori specializzati possono essere di aiuto nel miglioramento delle anomalie comportamentali o delle difficoltà cognitive. I maschi con malattia di Norrie possono aver bisogno di varie tipologie di assistenza da parte delle loro famiglie, amici e di chiunque se ne prenda cura, ma possono vivere una vita ricca e gratificante. Per ulteriori informazioni potete liberamente contattare la Norrie Disease Association (NDA) o uno dei professionisti facenti parte del nostro network e indicati nel retro della brochure.
Storia
Nel 1961, un danese, oculista di nome Mette Warburg ha riferito di una famiglia danese che ha mostrato sette casi di ereditariamalattia degenerativa tutto sette generazioni. Il primo membro della famiglia ad essere studiato a fondo è un ragazzo di 12 mesi.All’esame del bambino a tre mesi, si è notato che era normale, tranne che il suo obiettivo sembrava essere opaco e le sue iridi dura. [ 8 ] La zona dietro il suo obiettivo è stato riempito con una massa crescente giallastro. Cinque mesi più tardi, l’occhio sinistro è stato rimosso a causa di sospetti di retinoblastoma , un tumore canceroso sulla retina. Un istologico esame risulta un emorragicanecrotico massa nella camera posteriore , circondato da indifferenziata (immaturo, allacciamenti)
tessuto gliale . La diagnosi comprendeva una pseudo iperplasia della retina, ciliare e iride, pigmento dell’epitelio , ipoplasia e necrosi dello strato interno della retina, cataratta, e tisi bulbare . Questo significa che il suo occhio è stato rimosso perché il medico sospetta un tumore, anche se è emerso che era un difetto di sviluppo che ha portato alla malformazione parti interne dell’occhio. Poiché l’occhio non era funzionale, le cellule già cominciato a morire (necrosi) e il globo oculare ha cominciato a ridursi a causa della sua disfunzione (phthisi bulbi). In questa famiglia danese, cinque delle sette persone in questi casi sviluppato sordità più tardi nella vita.Inoltre, in quattro dei sette, capacità mentale era determinato a essere bassa. Dopo Warburg studiato letteratura in varie categorie mediche, ha scoperto 48 casi simili che si credeva fossero la causa da questa malattia pure. [ 8 ] Ha poi suggerito questa malattia è chiamato dopo un altro famoso oculista danese, Gordon Norrie (1855-1941). Norrie fu molto riconosciuto per il suo lavoro con i non vedenti e per essere un chirurgo presso l’Istituto danese per i ciechi per 35 anni. [ 9 ]
RICERCA
La ricerca continua a rivelare i modi in cui queste mutazioni genetiche causano le caratteristiche cliniche della malattia di Norrie, oltre a spiegare i fattori biologici che portano alla cecità, alla perdita uditiva, ai caratteristici aspetti cognitivi e comportamentali. Con una migliore comprensione di questi meccanismi, speriamo di capire come poter migliorare la prevenzione o il trattamento dei sintomi clinici e di dare supporto ai pazienti e alle famiglie colpite dalla Norrie. La forte somiglianza tra la ND e la FEVR ha portato a scoprire che i prodotti dei geni associati (NPD, FZD4, LRP5, TSPAN 12)interagiscono reciprocamente in un cosiddetto “percorso WNT canonico”. Mutazioni in uno di questi geni causano difetti nella vascolatura sanguigna, che sembra essere il meccanismo comune alla base dei differenti sintomi clinici.
|
|
|
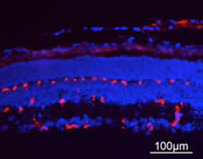
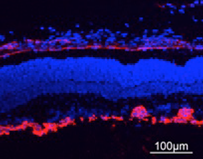
Sezioni attraverso la retina di un topo sano (a sinistra) e il mouse Norrie (a destra), che mostra la differenza nel sistema di vasi sanguigni (in rosso)
1. ^ Jump up to:a b c d e f g h i j k l m Sims, Katherine. “NDP-Related Retinopathies”. GeneReviews. Retrieved 28 January 2007.
2. ^ Jump up to:a b Halpin C, Owen G, Gutiérrez-Espeleta G, Sims K, Rehm H (2005). “Audiologic features of Norrie disease”. Ann Otol Rhinol Laryngol 114 (7): 533–8. PMID 16134349.
3. Jump up^ Dickinson JL, Sale MM, Passmore A, FitzGerald LM, Wheatley CM, Burdon KP, Craig JE, Tengtrisorn S, Carden SM, Maclean H, Mackey DA (2006). “Mutations in the NDP gene: contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity”. Clin Experiment Ophthalmol. 34 (7): 682–8. doi:10.1111/j.1442-9071.2006.01314.x.PMID 16970763.
4. ^ Jump up to:a b c “Norrie Disease”. Genetics Home Reference. Retrieved28 January 2007.
5. Jump up^ “NDP Norrie disease (pseudoglioma) [ Homo sapiens (human) ]”. Gene (National Center for Biotechnology Information). Retrieved 26 October 2013.
6. ^ Jump up to:a b c “Norrie disease (pseudoglioma)”. Genetics Home Reference. U.S. National Library of Medicine. March 2008. Retrieved 18 March 2008.
7. Jump up^ Meitinger T, Meindl A, Bork P, Rost B, Sander C, Haasemann M, Murken J (1993). “Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure”.Nature Genetics 5 (4): 376–380. doi:10.1038/ng1293-376.PMID 8298646.
8. ^ Jump up to:a b Warburg M (1961). “Norrie’s disease: a new hereditary bilateral pseudotumour of the retina”. Acta Ophthal. (Copenh)39: 757–772.
9. Jump up^ Gordon Norrie at Who Named It?. Retrieved 13 February 2007.
Bibliografia[modifica | modifica wikitesto]
Warburg M (1961). “Norrie’s disease: a new hereditary bilateral pseudotumour of the retina”. Acta Ophthal. (Copenh) 39: 757–772.
Cao A, Della Piccola B, Notarangelo LD, Malattie genetiche. Molecole e geni. Diagnosi, prevenzione e terapia, Piccin-Nuova Libraria editore, 2004, pp. 742, 766. ISBN 8829916528
Halpin C, Owen G, Gutiérrez-Espeleta G, Sims K, Rehm H (2005). “Audiologic features of Norrie disease”. Ann Otol Rhinol Laryngol 114 (7): 533–8. PMID 16134349.
Dickinson JL, Sale MM, Passmore A, FitzGerald LM, Wheatley CM, Burdon KP, Craig JE, Tengtrisorn S, Carden SM, Maclean H, Mackey DA (2006). “Mutations in the NDP gene: contribution to Norrie disease, familial exudative vitreoretinopathy and retinopathy of prematurity”. Clin Experiment Ophthalmol. 34 (7): 682–8. doi:10.1111/j.1442-9071.2006.01314.x. PMID 16970763.
Sims, Katherine. “NDP-Related Retinopathies.” Gene Reviews. 08 Aug 2006. 28 Jan 2007 <http://www.geneclinics.org/profiles/norrie/>
Norrie’s disease: a prospective study of development
H M GOODYEAR, P M SONKSEN, AND H McCONACHIE Department of Developmental Paediatrics, The Wolfson Centre, Hospital for Sick Children, London in Archives of Disease in Childhood, 1989, 64, 1587-1592
Una nuova ricerca può aiutare il trattamento della malattia di Norrie e familiare vitreoretinica essudativa (FEVR) New research may help treatment of Norrie disease and familial exudative vitreoretinopathy (FEVR)
Pubblicato il 22 Marzo 2004 alle 03:37 · 1 Comment
I ricercatori hanno scoperto che le mutazioni genetiche alla base di due disturbi oculari ereditarie sorgono in diverse componenti di un unico percorso di segnalazione intracellulare che è responsabile per lo sviluppo dei vasi sanguigni negli occhi.
Capire di più su come funziona questo pathway potrebbero fornire informazioni utili per lo sviluppo di farmaci per il trattamento delle due malattie. Tale informazione potrebbe anche aiutare a comprendere retina disturbi dei vasi sanguigni associate al diabete, degenerazione maculare, e nascita prematura.
Howard Hughes Medical Institute (HHMI) ricercatore Jeremy Nathans alla Scuola di Medicina dell’Università John Hopkins ha guidato il team di ricerca, che ha pubblicato i suoi risultati nel 19 MARZO 2004, numero della rivista Cell. Autori Co-lead dell’articolo erano HHMI associato Qiang Xu e specialista di ricerca HHMI Yanshu Wang. Altri autori sono dal National Institute on Deafness e altri disturbi della comunicazione, l’Università dello Utah, e l’Ospedale Wills Eye di Philadelphia.
I ricercatori hanno studiato due malattie ereditarie, malattie Norrie e essudativa familiare vitreoretinica (FEVR), il cui sottostante genetica difetti erano già note, ma il cui rapporto meccanicistico non era. Sindrome di Norrie, causata da un difetto nel gene per la proteina Norrin, produce cecità congenita e sordità progressiva dovuta al vaso sanguigno malformazioni nell’orecchio interno.”Quasi certamente la sequenza degli eventi all’interno dell’occhio è che vi è un problema in sviluppo vascolare, una crescita compensatoria dei vasi sanguigni, e leakiness nei vasi sanguigni che porta a cicatrici e infine cecità,” detto Nathans. La funzione della proteina Norrin era sconosciuta prima di questo nuovo lavoro, ha detto.
Il secondo disturbo i ricercatori hanno studiato, FEVR, “tende ad essere più mite e con una gamma di gravità tutta la strada da una modesta anomalia dei vasi che non pregiudica affatto la visione di una gravità di cicatrici che elimina completamente la visione”, ha detto Nathans. FEVR è conosciuto per derivare da difetti in un gene noto come Frizzled-4, che codifica per una proteina recettore (Fz4). Anche se Nathans ei suoi colleghi hanno studiato il Frizzled-4 gene e la proteina Fz4, non sapevano che cosa fosse segnale esterno, ha detto.
I primi suggerimenti che le due malattie possano essere funzionalmente collegati venuto quando i ricercatori hanno osservato somiglianze interessanti nei difetti dei vasi sanguigni nei due disturbi nei topi privi dei geni responsabili. Queste patologie dei vasi si sono verificati sia l’occhio e l’orecchio interno.
“Mentre c’era una somiglianza clinica, le due malattie sono stati tutt’altro che identici, perché i pazienti FEVR hanno una versione molto più mite del problema”, ha detto Nathans.Inoltre, ha detto, le persone con malattia di Norrie mostrano progressiva sordità, mentre quelli con FEVR no. “In speculando su come i pezzi del puzzle delle due malattie possano combaciare, abbiamo pensato che l’idea di un rapporto diretto sembrava sorta di folle – ma non troppo pazzo. Così, abbiamo deciso di provare un paio di esperimenti che potrebbero rivelare quel link. Nel giro di un mese abbiamo avuto la risposta. “
I ricercatori hanno condotto esperimenti di coltura cellulare utilizzando tecniche per tracciare l’interazione delle proteine, che ha rivelato che insieme le proteine Norrin e FZ4 attivare un percorso di sviluppo chiave denominata via di Wnt. I due componenti necessari anche un terzo co-recettore chiamato LRP5, che è noto per essere la chiave per la segnalazione Wnt, ha detto Nathans.
Gli esperimenti di coltura cellulare ha anche rivelato che la proteina Norrin era un trigger chiave per il recettore Fz4, selettivamente legame e attivarlo. “È importante sottolineare che abbiamo trovato che questo è molto alta affinità, altamente specifico vincolante”, ha osservato Nathans.
Inoltre, i ricercatori hanno studiato due forme umane di FEVR, trovando che nei pazienti con la malattia, mutazioni nel gene Frizzled-4 interferito con segnalazione Norrin-dipendente.
Se ridotta attività della via di segnalazione Norrin è infatti la causa di fondo dei due disturbi, Nathans ha detto, un farmaco che aumenta l’attività che potrebbe essere utile.Tale droga potrebbe anche essere efficace nel trattamento di malattie che si verificano nel corso della vita in cui il sistema Norrin-Fz4 potrebbe giocare un qualche ruolo, come il diabete e la degenerazione maculare legata all’età , Nathans aggiunto. Allo stesso modo, una condizione nota come retinopatia della prematurità, in cui i neonati prematuri trattati con alti livelli di ossigeno a causa della loro polmoni non sviluppate successivamente soffrono anormale sviluppo vascolare della retina, potrebbe anche essere beneficiato.
Dal momento che il percorso Norrin-Fz4 è specifico per gli occhi, un farmaco che manipola tale sistema potrebbe essere in grado di trattare questi disturbi senza molti effetti collaterali, un miglioramento significativo rispetto farmaci che più in generale influenzano la crescita vascolare, Nathans ha detto.
Tuttavia, ha sottolineato Nathans, molto più lavoro deve essere fatto per capire il ruolo della via Norrin-Fz4 in sviluppo vascolare. “Non siamo sicuri di quanto questo percorso generalizza,” ha detto. “In effetti, non sappiamo molto circa lo sviluppo vascolare in generale. È una domanda affascinante e provocatoria del perché la natura presi la briga di evolvere un percorso specifico solo per costruire queste navi l’occhio e l’orecchio interno. Si suggerisce che ci possono essere altri sistemi di sviluppo vascolare specializzati in altri tessuti e organi. “
Nuova Mutazione dei geni nella malattia di Norrie in pazienti giapponesi con malattia di Norrie e vitreoretinopatia familiare essudativa
1. Hiroyuki Kondo 1 ,
2. Minghui Qin 2 ,
3. Shunji Kusaka 3 ,
4. Tomoko Tahira 2 ,
5. Haruyuki Hasebe 4 ,
6. Hideyuki Hayashi 1 ,
7. Eiichi Uchio 1 e
8. Kenshi Hayashi 2
+Author Affiliations
- 1 Dal Dipartimento di Oftalmologia, Fukuoka University School of Medicine, Fukuoka, in Giappone; il
- 2 Divisione di analisi del genoma, Centro di Ricerca per l’informazione genetica, Istituto di Medicina di bioregolazione, Kyushu University, Fukuoka, in Giappone; il
- 3 Dipartimento di Oftalmologia, Osaka University Medical School, Suita, Giappone; e la
- 4 Dipartimento di Oftalmologia, Hiroshima Prefectural Hospital, Hiroshima, Giappone.
Astratto
SCOPO. Per cercare mutazioni nel gene malattia Norrie (NDP) in pazienti giapponesi con familiare vitreoretinica essudativa (FEVR) e la malattia Norrie (ND) e per delineare le caratteristiche cliniche mutazione associata.
METODI. sequenziamento diretto dopo la reazione a catena della polimerasi di tutti gli esoni del gene NDP è stato effettuato su sangue raccolto da 62 probandi (31 familiare e 31 simplex) con FEVR, da 3 probandi con ND, e da alcuni dei loro familiari. Sono stati valutati i sintomi e segni clinici nei pazienti con mutazioni. X-inattivazione nelle femmine portatrici stato esaminato in tre famiglie FEVR utilizzando leucociti DNA.
RISULTATI. Quattro nuove mutazioni-I18K, K54N, R115L, e IVS2-1G → A e uno riferito mutazione, R97P, nel gene NDP sono stati identificati in sei famiglie. La gravità di vitreoretinica variava tra questi pazienti. Tre probandi con o K54N o R115L avuto caratteristiche tipiche di FEVR, mentre i probandi con R97P aveva quelle di ND. Famiglie con IVS2-1G → A esposti sia caratteristiche ND o FEVR. Un probandi con I18K presentato con notevole eterogeneità fenotipica tra i due occhi. Inoltre, i vettori influenzato femminili in una famiglia la mutazione K54N presentato con diversi gradi di anomalie vascolari nella periferia della retina. Profili X-inattivazione indicato che l’inclinazione non era significativamente differente tra le donne colpiti e non colpiti.
CONCLUSIONI. Queste osservazioni indicano che mutazioni del gene NDP possono causare ND e il 6% dei casi FEVR nella popolazione giapponese. Il saggio X-inattivazione con leucociti non può essere predittiva della presenza di una mutazione in donna portatori colpiti.
Sezione precedenteSezione successiva
Introduzione
Sindrome di Norrie (ND) è una rara, X-linked malattia recessiva caratterizzata da cecità congenita a causa di masse retrolental denominato pseudogliomas. 1 ritardo mentale e la perdita dell’udito si osservano anche in circa il 25% dei casi. 1 ND è geneticamente omogenea e è causata da mutazioni nel gene NDP, che codifica per la proteina Norrin 133 aminoacidi. 2 3 Questa proteina non ha forti identità di sequenza con altre proteine note, ma i confronti sequenza e studi di modellazione hanno previsto che ha un numero regolare e la distanza di circa sei residui mezza cistina e alcuni residui idrofobici per formare domini nodo cistina. 4 La struttura terziaria di Norrin ha una forte somiglianza con fattore di crescita trasformante-β.5 Sebbene l’esatta funzione della proteina è sconosciuta, è stato suggerito a svolgere un ruolo nello sviluppo e la regolamentazione del neuroectoderma. 4 6
Familial vitreoretinopatia essudativa (FEVR) è una malattia geneticamente eterogenea vitreoretinica caratterizzata da un deficit nello sviluppo vascolare della retina periferica che porta a varie complicazioni secondarie, tra cui la trazione maculare, il distacco della retina, e pieghe retiniche. 7 8 9 La malattia ha diversi gradi di gravità, che vanno dall’assenza di sintomi visivi di totale cecità. 10 FEVR ha tre modelli di eredità:. autosomica dominante (adFEVR), autosomica recessiva (arFEVR), e X-linked recessiva (XFEVR) 7 10 11 12 Ad- e arFEVR sono conosciuto per causare difetti di entrambi i coreceptors Wnt Frizzled-4 o lipoproteine a bassa densità proteina recettore-like 5 (LRP5) che attiva la classica via di segnalazione Wnt. 13 14 15 16 Questo percorso svolge un ruolo centrale nello sviluppo vascolare della retina. 17 XFEVR è causato anche da Norrin mutante, 18 e Norrin stesso è stato recentemente trovato ad agire come un ligando per frizzled-4. 17 Ciò indicherebbe un meccanismo molecolare comune alla base sia ND e FEVR.
Un gran numero di mutazioni nel gene NDP sono stati descritti: traslocazione e inversione mutazioni, diverse mutazioni per delezione, e più di 80 mutazioni puntiformi (per la revisione, vedi Rif. 19 ). 20 diverse alterazioni strutturali in Norrin possono portare a diversi gradi di fenotipica severità. 19 di eliminazione e di troncamento mutazioni in NDP causa ND senza eccezione, mentre le mutazioni missense causano sia FEVR o ND. 19 mutazioni che non interrompono eventuali legami disolfuro previsti sono più propensi a esprimere fenotipi più lievi. 4 19 20 2122 Tuttavia, solo alcuni studi sperimentali hanno affrontato l’importanza funzionale di mutazioni di senso e la loro correlazione con la gravità. 23 Inoltre, non sono stati riportati studi sulla se le mutazioni possono prevedere la gravità della malattia. 24 Alcuni mutazioni di senso sono noti per essere associati con altre malattie della retina, come la retinopatia del prematuro e malattie Coats. 25 26 27
Così, uno stabilimento di una relazione genotipo-fenotipo in grado di offrire informazioni aggiuntive che possono portare ad una prognosi più precisa, la diagnosi prenatale e la consulenza genetica. Per quanto a nostra conoscenza, non ci sono segnalazioni relative a mutazioni in NDP in pazienti giapponesi con FEVR.Abbiamo quindi studiato le coorti di pazienti giapponesi con FEVR e ND dallo screening NDP e mutazioni identificate in pazienti con FEVR e ND. Abbiamo anche osservato femmine portatrici che presentano caratteristiche FEVR raggruppati in una sola famiglia, e abbiamo esaminato il profilo della inattivazione del cromosoma X nelle femmine portatrici utilizzando leucociti DNA.
Sezione precedenteSezione successiva
Metodi
Partecipanti
Sessantadue probandi (31 familiari e 31 simplex) con FEVR, 3 probandi con ND, e alcuni dei loro familiari sono stati esaminati presso il Dipartimento di Oftalmologia presso l’Università di Fukuoka o Osaka University. Un consenso informato firmato è stato ottenuto da tutti i soggetti, e le procedure utilizzate conforme ai principi della Dichiarazione di Helsinki. Inoltre, il protocollo sperimentale è stato approvato dal Ethics Review Board di Fukuoka University.
Tutti i pazienti erano giapponesi e sono nati a termine di peso normale. La diagnosi di FEVR era basata sulla presenza di almeno uno dei segni clinici tipici, (ad esempio, periferiche avascularization retinica con formazione anormale retinica vascolare, gravi essudati retinici, neovascolarizzazione della retina, la massa fibrovascular periferico, ectopia maculare, pieghe retiniche, distacco della retina , e emorragia del vitreo) La diagnosi di ND è stata fatta per la presenza di distacco di retina bilaterale o retinica pieghe con tessuto fibroso retrolental, cecità entro il primo anno di vita, il 24 la presenza o l’assenza di ritardo mentale e perdita di udito, e una storia familiare di X-linked recessive ereditarietà.
Esami clinici
Tutti i pazienti hanno avuto un esame oftalmologico completo, che includeva misure Snellen acutezza visiva, la pressione intraoculare, con lampada a fessura biomicroscopia, e oftalmoscopia. Alcuni pazienti sono stati esaminati anche da angiografia e / o ecografia fluoresceina. La densità minerale ossea (BMD) è stata valutata in alcuni casi adulti misurando l’area della BMD della colonna lombare con Mineralometria ossea computerizzata (modello QDR-4500A, Hologic Inc., Waltham, MA). Per le analisi statistiche, il valore di BMD è stato convertito in un punteggio SD (z -score) adatto per soggetti di controllo al genere e di pari età.
Studi di laboratorio
Campioni di DNA sono stati estratti dal sangue periferico con un kit di estrazione del DNA (QIAamp, Qiagen, Valencia, CA). I primer che tra parentesi tutti gli esoni di NDP usati erano 5′-attGGCACTTTTCCATTTGACA-3 ‘e 5′-gttATGCTCGGTTTGGAAAGAAG-3′ per esone 1; 5’-attGGATCCTAGGAGGTGAAGCC-3 ‘e 5′-gttCTGAGGGAAATGCTCTCCT-3′ per esone 2; e 5’-attATGCCCACAGAGTAACCACC-3 ‘e 5′-gttCATCCAGAAGCCACACACAG-3’ per esone 3. sequenze di att o GTT Tagged sono stati aggiunti alla fine del 5 ‘a fini postlabeling. 28
Reazione a catena della polimerasi (PCR) e sequenziamento sono stati eseguiti.Temperature di ricottura per PCR sono stati 60 ° C per esone 1 e 65 ° C per esoni 2 e 3. Altri dettagli delle procedure sono state descritte. 14 28
Un inattivazione del cromosoma X è stata determinata da CAG ripetizioni polimorfiche del gene del recettore degli androgeni umano che sono stati amplificati dal DNA del sangue periferico. 29 Lo stato di metilazione dei due cromosomi X di ogni donna è stato determinato utilizzando metilazione-sensibile enzima di restrizione (Hpa II , New England Biolabs, Beverly, MA). Sequenze originali dei primer sono stati utilizzati, ma con il tag “att” o “GTT” alla fine del 5 ‘per la fluoresceina etichettatura degli amplificati. 28 Gli ampliconi marcati sono stati separati mediante elettroforesi (modello 310 sequencer, Applied Biosystems [ABI], Foster City, CA). Un’analisi dimensioni frammento è stato quindi eseguito (software GeneScan; ABI) i segnali provenienti dai due cromosomi di ciascuna donna sono stati regolati prima digestione con enzima metilazione sensibile, e quindi un rapporto segnale relativo del cromosoma interessato è stata calcolata dopo la digestione.
Sezione precedenteSezione successiva
Risultati
Analisi Mutazione
Il DNA da 62 FEVR e tre ND pazienti sono stati analizzati per mutazioni in tre esoni e fiancheggiano i confini esone-introne del gene NDP. Cinque differenti mutazioni sono state identificate in quattro (tre simplex e un familiare) pazienti FEVR e due pazienti con ND (Tabella 1) . Le tre nuove mutazioni erano una T → A transizione, c.53 T → A in esone 2, con un conseguente cambiamento isoleucina-to-lisina a codone 18 (I18K) in famiglia 1; una transizione G → C, c.162G → C, nell’esone 2, con conseguente cambiamento lisina-to-asparagina a codone 54 (K54N) nei pazienti con FEVR nelle famiglie 2 e 3; una trasversione G → T, c.344G → T, in esone 3, con un conseguente cambiamento arginina-a-leucina a codone 115 (R115L) in un paziente con simplex FEVR in famiglia 4; e una G → A trasversione, IVS2-1G → A al sito accettore dell’esone 3 in un paziente con ND in famiglia 6. One precedentemente riportato mutazione, una transizione G → C, c.290G → C, nell’esone 3, con conseguente in un cambiamento nonconserved arginina-a-prolina a codone 97 (R97P), è stato trovato in un paziente con ND in famiglia 5. Tutti probandi erano uomini, e quindi questi cambiamenti sono stati interpretati come emizigote. Questi cambiamenti di sequenza non sono stati trovati in 180 individui non imparentati e non affetti (tutti erano femmine) nella popolazione giapponese.
Visualizza questa tabella:
T GRADO 1.
Le mutazioni nel gene NDP e risultati clinici associati
Quando allineamento di sequenze multiple di Norrin umana è stata effettuata tra le sequenze di specie disponibili, codoni I18, K54, R97, R115 e sono stati conservati in ratti e topi (dati non riportati). K54N, R97P, e R115L erano mutazioni nonconserved e si trovavano all’interno del dominio cistina nodo. I18K era una mutazione nonconserved e si trovava all’interno della sequenza del segnale N-terminale. L’effetto preciso della mutazione splicing non è stato determinato per l’espressione di Norrin è limitata alla retina e cervello tessuti, 2 3 e tale RNA era disponibile dai pazienti. Tuttavia, i criptici siti accettore splicing a 17- e 125-bp a valle dei siti originali sono stati desunti da una analisi computazionale (GeneSplicer, http://www.cbcb.umd.edu/software/GeneSplicer/, fornito da pubblico dominio dal L’Università del Maryland Centro di Bioinformatica e Biologia Computazionale, College Park, MD). Entrambe le alternative splicing porterebbe ad una delezione 17 e 125-bp all’inizio dell’esone 3, risultante in un frameshift dopo codoni 64 e 100, rispettivamente, seguito da un terminale allungata.
I campioni di membri della famiglia erano disponibili per tutti i casi, e una cosegregazione di questi cambiamenti sequenza è stata cercato dopo il sequenziamento. Tutte le modifiche cosegregate come la forma recessiva X-linked della malattia in tutte le famiglie, tranne per la famiglia 3. In questa famiglia, la madre e tre sorelle erano eterozigoti per K54N. Hanno presentato con molto diversi gradi di anomalia vascolare, e la madre e una sorella ha avuto una diagnosi di FEVR. Perché la nonna materna e il nonno non ha effettuato K54N, una mutazione de novo è stato considerato presente nella madre.
Fenotipi dei pazienti con FEVR
I18K.
Paziente III-1 a famiglia 1 (Fig. 1) è stato un bambino di 6 mesi. E ‘nato a termine di peso normale. Quando è stato esaminato a 4 mesi di età, massa retrolental con totale distacco di retina è stata trovata nell’occhio destro. Le caratteristiche tipiche di FEVR, come periferiche avascularization temporale alla macula e neovascolarizzazione dei vasi retinici, sono stati osservati nell’occhio sinistro in cui era stata effettuata la fotocoagulazione laser (Fig. 2A) . Nessun membro della famiglia aveva FEVR, ma la madre era eterozigoti per la mutazione I18K.
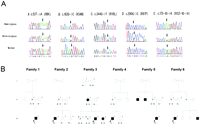
Visualizza versione più grande:
F IGURA 1.
Cromatogrammi e pedigree di sei famiglie con FEVR o ND. (A)Le mutazioni nei geni NDP in pazienti con FEVR o ND: Frecce.posizioni dei nucleotidi alterati.(B) pedigree di sei famiglie che illustrano la cosegregazione delle mutazioni NDP in FEVR famiglie 1 a 4 e in ND famiglie 5 e 6. simboli solidi: gli individui con una diagnosi di FEVR o ND: Frecce. Probandi. Gli individui da cui sono stati ottenuti i dati di sequenza sono indicati con i numeri: A, B, C, D, ed E indicano la sequenza cambia I18K, K54N, R115L, R97P e IVS-1G →, rispettivamente A, che sono anche sopra indicato i dati di traccia nella parte superiore. N indica una sequenza wild-type. * Una diagnosi di FEVR non è stato fatto perché è stata osservata solo tortuosità minima dei vasi retinici.
Visualizza versione più grande:
F IGURA 2.
Fotografie fondo e angiografia con fluoresceina dei pazienti.(A) fluoresceina angiogramma dell’occhio sinistro della probanda della famiglia 1 (III-1), che mostra neovascolarizzazione e una grave avascularization retinica tipica di FEVR. Immagine (B)Fundus del diritto occhio del probando della famiglia 2 (III-1), che mostra una macula trascinato con persistente residuo hyaloid. (C) occhio destro della probanda della famiglia 3 (III-3) mostra una ricerca simile a quello mostrato in (B). Immagine (D) fondo dell’occhio sinistro della probanda della famiglia 4 (III-1) mostra un distacco di retina totale. (E)fotografia esterna l’occhio destro di probando della famiglia 5 (III-1) che mostra fibroplasia retrolental. (F) fotografia esterno dell’occhio sinistro della probanda della famiglia 6 (III-1) che mostra una camera anteriore piatta con opacità corneale progressione di buphthalmia.
K54N.
Paziente III-1 in famiglia 2 (Fig. 1) è stato un bambino di 6 anni che aveva pieghe retiniche bilaterali simile persistente iperplastico primaria del vitreo (PHPV). Questa condizione ha progredito per distacco della retina e la trazione maculare con avascularization temporale nell’occhio sinistro (Fig. 2B) . Aveva subito un intervento chirurgico del vitreo nell’occhio sinistro, ma la retina non poteva essere riattaccato. I suoi acuità visiva corretti erano 0,5 OD e movimento della mano OS.Nessun membro della famiglia aveva FEVR, ma la madre era eterozigoti per la mutazione K54N.
Paziente III-3 della famiglia 3 era un ragazzo di 5 anni che ha avuto FEVR con tortuosità vascolare retinica e avascularization della retina temporale periferica. I cambiamenti nel posteriore della retina vitreo e simili a quelle del probando della famiglia 2 (Fig. 2C) . Profilattica fotocoagulazione retinica e una fibbia circondare sono stati posti su entrambi gli occhi. La madre e una sorella aveva periferici anomalie vascolari retiniche compatibili con FEVR (Figg. 3A 3B) , e le altre due sorelle avevano una forma più lieve di tortuosità vascolare (Fig. 3C 3D) .
Visualizza versione più grande:
F IGURA 3.
Immagini fondo e angiografia con fluoresceina degli occhi dei portatori femminili in famiglia 3. fluoresceina angiografia della madre (A) e la sorella III-4 (B)che mostra i segni tipici di FEVR, come avascularization e tortuosità dei vasi retinici. Le altre due sorelle III-1 (C) e III-2(D) hanno mostrato minimo tortuosità vaso retinico senza avascularization.
R115L.
Paziente III-1 in famiglia 4 era un uomo di 21 anni che aveva una massa fibrovascolare adiacente ai terminali del avascularization retina bilateralmente. Alla sua prima visita al nostro ospedale all’età di 10 anni, il suo migliore acuità visiva corretta era 1.0 con miopia elevata in entrambi gli occhi. A causa della grave trazione vitreoretinica dal temporale massa fibrovascular alla macula, chirurgia della retina profilattico con sclerale deformazione e fotocoagulazione laser è stata effettuata su entrambi gli occhi, all’età di 16 anni. Successivamente, un distacco della retina totale sviluppato nell’occhio sinistro (Fig. 2D) . Nonostante i ripetuti interventi chirurgici, la retina è rimasto distaccato, e l’olio di silicone è rimasto negli occhi. L’esame oculare della madre e del fratello non ha rivelato alterazioni retiniche.
Fenotipo di pazienti con ND
R97P.
Paziente III-1 in famiglia 5 era un ragazzo 1-year-old. E ‘nato a termine di peso normale. Leucocoria bilaterale è stato notato a 2 mesi di età. Un esame ha mostrato una camera anteriore piatta e una massa retrolental con distacco di retina totale in entrambi gli occhi. ND è stato fortemente sospettato (Fig. 2E) .Lensectomia stata eseguita per creare e mantenere la camera anteriore. Vision è stato limitato alla percezione della luce in entrambi gli occhi, e lui non riusciva a seguire un dito che si muove con entrambi gli occhi. Non aveva un ritardo di sviluppo o mentale, e il suo udito era normale. Due zii materni sono stato cieco da quando erano neonati, e uno di loro è mentalmente ritardato.
IVS2-1G → A.
Paziente III-1 in famiglia 6 era un ragazzo 1-year-old. Egli è stato presentato al nostro ospedale a 3 mesi di età, e ND era sospettato a causa di tessuto fibroso retrolental e distacco della retina con una camera anteriore piatta in entrambi gli occhi. Il buphthalmia progredito nell’occhio sinistro (Fig. 2F) , e lensectomia stata eseguita per preservare la camera anteriore in entrambi gli occhi. Sensazione di luce è stata conservata, ma non riusciva a seguire un oggetto in movimento. Anche se entrambi i genitori non avevano anomalie oculari, nipote materno aveva una diagnosi di FEVR causa di pieghe retiniche bilaterali a 1 anno di età.
X-inattivazione Assay
Tre donne in famiglia FEVR con la mutazione K54N sono stati analizzati per X-inattivazione. Il rapporto segnale relativo di entrambi i cromosomi della madre colpita in famiglia 3 non era ovviamente distorta a 37%: 63%. Le due sorelle che portano la mutazione K54N eterozigoti (III-1: inalterato e III-4: colpiti) erano omozigoti. Abbiamo provato altri marcatori polimorfici per un saggio X-inattivazione base-PCR. 30 , nonostante le condizioni variabili per l’amplificazione testato, nessun prodotto è stato ottenuto, e lo stato di X inattivazione non è stato possibile determinare. Altri due femmine portatrici non affetti nelle famiglie 2 e 4 sono stati anche analizzati. Poiché questi casi erano simplex, non vi era alcuna possibilità di determinare quale dei cromosomi X potrebbe essere portare le mutazioni. I rapporti dei segnali relativi sono stati il 16%: 84% in famiglia 2 e il 27%: il 73% in famiglia 4.
In conclusione, è stata osservata alcuna differenza significativa di inclinazione tra donne colpiti e non colpiti.
Sezione precedenteSezione successiva
Discussione
Abbiamo identificato cinque mutazioni nel gene NDP in sei famiglie. Uno era una mutazione splicing romanzo, e gli altri quattro erano missenso cambiamenti. Tre di queste mutazioni missense erano nuova, e loro si trovavano all’interno del dominio cistina nodo o segnale sequenza N-terminale. Tutte le mutazioni segregati con la malattia e non sono stati trovati in individui normali da parte della popolazione giapponese. Questi risultati suggeriscono che queste mutazioni sono patogeni. I nostri dati indicano che le mutazioni NDP causano il 6% (4/62) del FEVR nella popolazione giapponese.
Due pazienti con nuova mutazioni, K54N e R115L, hanno avuto i risultati tipici della FEVR. Da segnalare, due non imparentati probandi con la mutazione K54N avuto un guasto nella regressione del sistema vascolare hyaloid. Questo risultato è coerente con quelli in un modello animale con una mutazione NDP. 31
Finora, le mutazioni che causano FEVR si limitano ai residui 41-58 e 121-126, così come residuo 110. 32 33 34 35 36 Dalla struttura secondaria prevista, queste regioni sono all’interno del componente principale del dominio cistina nodo :. primo filone beta con conseguente ciclo di residui 41-58, e il quarto filone beta per i residui 121-126 5 Il residuo di cisteina 110 è direttamente coinvolto nel legame disolfuro-dimero formando. 5 mutazioni in queste regioni possono agire interrompendo il folding delle proteine o interferendo direttamente con interazioni Norrin-recettore. 5 37 K54N e R115L abbinati con una tale predisposizione, e lo spettro mutazionale è stato ampliato. Sebbene l’importanza funzionale di ciascuna mutazione debba essere esaminata in maggior dettaglio, una predisposizione in una regione specifica può essere coinvolta nella patogenesi di FEVR.
Contrariamente a K54N e R115L, la nuova mutazione I18K aveva una funzione diversa. Questa mutazione era situato all’interno della sequenza del segnale N-terminale. Sequenze segnale giocano un ruolo nell’inserimento delle proteine secretorie nella membrana. La caratteristica di sequenze segnale è una regione core idrofobico che è affiancato sul lato C-terminale di una regione polare con una carica netta positiva. 38 Una sostituzione del isoleucina aminoacido polare per la base lisina altererebbe le proprietà fisico-chimiche del segnale sequenza putativa in modo significativo e può impedire un efficiente trasporto extracellulare di Norrin. Finora, solo il fenotipo ND-simile è nota per essere associata con mutazioni nella sequenza segnale putativa, mentre la FEVR-fenotipo non è. 39 40
Clinicamente, i probandi con I18K avuto una diagnosi di FEVR causa del tipico avascularization retina nell’occhio sinistro. Tuttavia, l’occhio collega esposto una gravità diversa, con un distacco di retina totale infantile. È possibile che le mutazioni nella sequenza segnale causano FEVR nonché ND.
La mutazione ricorrente, R97P, espone il fenotipo di ND, e questa mutazione è stata trovata in una famiglia con una storia di cecità infantile e ritardo mentale, che è coerente con la precedente relazione di ND. 41 Codone 97 è adiacente a uno dei mezza residui di cistina (C96) e partecipa alla formazione dei legami disolfuro. Così, R97P può interferire con il legame disolfuro. La mutazione splicing causato un fenotipo più variabile. Il probando presentato con le caratteristiche tipiche di ND, mentre un nipote materno aveva una diagnosi di FEVR.
Nonostante il fatto che ND e FEVR sono viste come malattie distinte, lo spettro fenotipico di ciascuno è diversa, e condividono molte caratteristiche comuni, come microftalmia, cataratta, pieghe della retina, e risultati PHPV-like. 42 43 Pertanto, Allen et al . 44 recentemente proponevano che le malattie mutazione associataNDP dovrebbero essere chiamati ND perché il termine FEVR non adeguato a trasmettere il potenziale spettro della gravità della malattia. È stato dimostrato che la mutazione K58N, che è molto simile alla mutazione K54N trovato in questo studio, può portare a uno o FEVR ND. 35 45 Intrafamilial variabilità del fenotipo oculare è stato anche descritto, e il coinvolgimento di ulteriori (epigenetica o) fattori genetici stato suggerito. 46 Questi risultati, nonché la mutazione splicing in questo studio implica il valore prognostico limitato di test genetici. I medici dovrebbero essere consapevoli del fatto che la variabilità fenotipica in ND / FEVR rende spesso la diagnosi clinica iniziale inadeguato, e la consulenza genetica può essere compromessa. 1
Anche se è raro, sia ND e XFEVR possono verificarsi nelle donne. 46 47 48 49 La nostra relazione è il primo a dimostrare che le femmine portatrici colpite espositrici FEVR sono stati raggruppati in una sola famiglia. Infatti, la malattia in questa famiglia è stato inizialmente considerato come dominante ereditata.Rispetto ai risultati della retina del probando maschile, i fenotipi di questi vettori sono stati molto più mite, che vanno dai livelli subnormali con tortuosità minima dei vasi retinici di una tipica forma di degenerazione della retina periferica.
Nei disturbi recessivi X-linked, le donne eterozigoti possono essere colpiti perché hanno il cromosoma X che non portano la mutazione come quella inattiva nella maggior parte delle loro celle. Questo processo è chiamato inclinazione di X inattivazione. 50 51 Le mutazioni NDP in femmine portatrici colpiti sono stati attribuiti a questo meccanismo. 46 47 48 49 Abbiamo quindi analizzato se l’X-inattivazione delle donne colpite è stata distorta a favore del fenotipo. Confrontando le altre femmine portatrici non affetti, X-inattivazione nella madre colpita non era apparentemente distorta mentre quelle sorelle colpiti non poteva essere determinato. Shastry et al. 49 testato un paziente con ND trasportano la mutazione NDP e trovò una discordanza di X-inattivazione a favore del cromosoma interessato, che è stato attribuito ad un modello di espressione presumibilmente diverso nei leucociti da quello in tessuto retinico. I nostri risultati possono mostrare che il saggio X-inattivazione di leucociti non fornisce informazioni sufficienti per prevedere il fenotipo nelle femmine portatrici. Il raggruppamento dei pazienti di sesso femminile affetti all’interno di una famiglia non era facilmente attribuibile a eventi indipendenti di skewed X-inattivazione.Inoltre, i dati recenti suggeriscono che la regione genomica compresi NDP a Xp-ter per Xp.12 rischiava di sfuggire parzialmente X inattivazione. 52 Ulteriori indagini sono necessarie per capire perché FEVR e ND sviluppati nei pazienti di sesso femminile affetti.
In sintesi, abbiamo identificato cinque mutazioni nel gene NDP che ha portato in FEVR o ND. Allo stato attuale, i risultati dei test genetici devono essere usati con cautela quando offre una prognosi o di consulenza. Ulteriori studi per identificare le correlazioni fenotipo-genotipo, nonché esplorare la rilevanza funzionale di ogni mutazione aiuteranno a capire il meccanismo di questi disturbi e la variabilità clinica.
Sezione precedenteSezione successiva
Sezione precedenteSezione successiva
Ringraziamenti
Gli autori ringraziano i pazienti e le loro famiglie.
Sezione precedenteSezione successiva
Note
- Supportato da Grant-in-Aid per la Ricerca Scientifica (C) 15.591.883 e 18.591.944.
- Presentato per la pubblicazione 4 settembre 2006; revisione 17 ottobre 2006;accettato 26 dicembre 2006.
- Disclosure: H. Kondo, None; M. Qin, None; S. Kusaka, None; T. Tahira, None;H. Hasebe, None; H. Hayashi None; E. Uchio, None; K. Hayashi, None
- Le spese per la pubblicazione di questo articolo sono stati finanziati in parte dal pagamento di carica pagina. Questo articolo deve essere contrassegnato“advertisement” secondo 18 USC §1734 esclusivamente per indicare questo fatto.
- Corrispondente autore: Hiroyuki Kondo, Dipartimento di Oftalmologia, Fukuoka University School of Medicine, 7-45-1, Nanakuma, Jonanku, Fukuoka 814-0180, Giappone; hkondo @ fukuoka-u.ac.jp.
· Copyright 2007 L’Associazione
Riferimenti
. Warburg malattia di M. Norrie difetti di nascita Orig Artic Ser . 1971 ; 7 :117 -124.Medline
Chen ZY, Hendriks RW, Jobling MA, et al. Isolamento e caratterizzazione di un gene candidato per Norrie malattia. Nat Genet . 1992 ; 1 : 204 -208.CrossRefMedlineWeb of Science
Berger W, Meindl A, van de Pol TJ, et al. Isolamento di un gene candidato per la malattia di Norrie dalla clonazione posizionale. Nat Genet . 1992 ; 2 :84 .MedlineWeb of Science
Meindl A, Berger W, Meitinger T, et al. Norrie malattia è causata da mutazioni in una proteina extracellulare simile C-terminale del dominio globulare mucine. Nat Genet . 1992 ; 2 : 139 -143.
Meitinger T, Meindl A, Bork P, et al. Modellistica molecolare della proteina malattia Norrie prevede un fattore di crescita struttura terziaria cistina nodo.Nat Genet . 1993 ; 5 : 376 -380.CrossRefMedlineWeb of Science
Chen ZY, Battinelli EM, Hendriks RW, et al. Norrie malattia genetica: caratterizzazione di delezioni e possibile funzione. Genomics . 1993 ; 16 :533 -535.CrossRefMedlineWeb of Science
Criswick VG, Schepens CL. Familial vitreoretinopatia essudativa. Am J Ophthalmol . 1969 ; 68 : 578 -594.
Tasman W, Augsburger JJ, Shields JA, Caputo A, Annesley WH, Jr. familiare vitreoretinica essudativa. Trans Am Ophthalmol Soc . 1981 ; 79 : 211 -226.Medline
Mukai S, Mukai E, Puliafito CA. Familiare vitreoretinica essudativa. Eds Berson EL D’Amico DJ Schepens CL. Principles and Practice of Ophthalmology. 1994 ; 813 -817. WB Saunders Philadelphia.
Canny CL, GL Oliver. Fluoresceina risultati angiografici a familiare vitreoretinica essudativa. Arch Ophthalmol . 1976 ; 94 : 1114 -1120.CrossRefMedlineWeb of Science
de Crecchio G, Simonelli F, Nunziata G, et al. . Autosomica recessiva familiare vitreoretinica essudativa: prova per eterogeneità genetica Clin Genet. 1998 ; 54 : 315 -320.CrossRefMedlineWeb of Science
Fullwood P, J Jones, Bundey S, et al. X legata vitreoretinopatia essudativa: caratteristiche cliniche e analisi di linkage genetico. Br J Ophthalmol . 1993 ;77 : 168 -170.Estratto / LIBERO testo completo
Robitaille J, MacDonald ML, Kaykas A, et al. Mutant frizzled-4 interrompe l’angiogenesi retinica nella familiare vitreoretinica essudativa. Nat Genet .2002 ; 32 : 326 -330.CrossRefMedlineWeb of Science
Kondo H, Hayashi H, K Oshima, Tahira T, Hayashi K. Frizzled 4 gene (FZD4) mutazioni in pazienti con familiare vitreoretinica essudativa con espressività variabile. Br J Ophthalmol . 2003 ; 87 : 1291 -1295.
Estratto / LIBERO testo completo↵
Toomes C, Bottomley HM, Jackson RM, et al. Mutazioni in LRP5 o FZD4 alla base della comune familiare essudativa locus sul cromosoma 11q vitreoretinica. Am J Hum Genet . 2004 ; 74 : 721 -730.CrossRefMedlineWeb of Science
Jiao X, Ventruto V, Trese MT, Shastry BS, Hejtmancik JF. Autosomica recessiva familiare vitreoretinica essudativa è associata a mutazioni in LRP5.Am J Hum Genet . 2004 ; 75 : 878 -884.CrossRefMedlineWeb of Science
Q Xu, Wang Y, Dabdoub A, et al. Sviluppo vascolare della retina e dell’orecchio interno: controllo Norrin e Frizzled-4, una coppia ligando-recettore ad alta affinità. cella . 2004 ; 116 : 883 -895.CrossRefMedlineWeb of Science
Chen ZY, Battinelli EM, Fielder A, et al. Una mutazione nel gene malattia Norrie (NDP) associata a X-linked familiare vitreoretinica essudativa. Nat Genet . 1993 ; 5 : 180 -183.CrossRefMedlineWeb of Science
Berger W, Ropers HH. Norrie malattia. Scriver CR Beaudet AL Sly WS Valle D eds. Le basi metaboliche e molecolari della malattia ereditaria . 2001 ; 5977-5985. McGraw Hill New York.
Riveiro-Alvarez R, Trujillo-Tiebas MJ, Gimenez-Pardo A, et al. Variazioni genotipo-fenotipo in cinque famiglie spagnole con malattia Norrie o X-linked FEVR. Mol Vis . 2005 ; 11 : 705 -712.MedlineWeb of Science
Joos KM, Kimura AE, Vandenburgh K, Bartley JA, Pietra EM. Reperti oculari associati a una mutazione Cys39Arg nella malattia gene Norrie. Arch Ophthalmol . 1994 ; 112 : 1574 -1579.CrossRefMedlineWeb of Science
Wong P, MacDonald IM, Sood R, et al. Identificazione e caratterizzazione parziale di un gene candidato per retinopatie X-linked con un approccio laterale. Genomics . 1993 ; 15 : 467 -471.CrossRefMedlineWeb of Science
Perez-Vilar J, Hill RL. Sindrome di Norrie proteine (Norrin) costituisce oligomeri disolfuro-linked legati alla matrice extracellulare. J Biol Chem .1997 ; 272 : 33410 -33415.Estratto / LIBERO testo completo
Royer G, Hanein S, Raclin V, et al. NDP mutazioni del gene in 14 famiglie francesi con Norrie malattia. Hum Mutat . 2003 ; 22 : 499 .CrossRefMedline
Nero GC, Perveen R, Bonshek R, et al. Malattia di Coats della retina (unilaterale telangiectasis retinica) causata da mutazione somatica nel gene NDP: un ruolo per Norrin nell’angiogenesi retinica. Hum Mol Genet . 1999 ; 8: 2031 -2035.Estratto / LIBERO testo completo
Shastry BS, Pendergast SD, Hartzer MK, Liu X, Trese MT. L’identificazione di mutazioni missense nel gene malattia Norrie associati retinopatia avanzata della prematurità. Arch Ophthalmol . 1997 ; 115 : 651 -655.
Hutcheson KA, Paluru PC, Bernstein SL, et al. Norrie sequenza del gene malattia varianti in una popolazione etnicamente diversificata con retinopatia della prematurità. Mol Vis . 2005 ; 11 : 501 -508.MedlineWeb of Science
Kondo H, Tahira T, H Hayashi, Oshima K, Hayashi K. microsatelliti genotipizzazione di post-PCR fluorescente marcatori. Biotecnologie . 2000 ;29 : 868 -872.MedlineWeb of Science
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. La metilazione di HpaII e HhaI siti vicino CAG repeat polimorfica del gene del recettore androgeno-umano è correlato con l’inattivazione del cromosoma X.Am J Hum Genet . 1992 ; 51 : 1229 -1239.MedlineWeb of Science
Hendriks RW, Chen ZY, Hinds H, Schuurman RK, Craig IW. Un cromosoma X inattivazione saggio basato sulla metilazione differenziale di un isola CpG accoppiato ad un polimorfismo VNTR all’estremità 5 ‘della monoamino ossidasi Un gene. Hum Mol Genet . 1992 ; 1 : 662 .Estratto / LIBERO testo completo
Rehm HL, Zhang DS, Brown MC, et al. Difetti vascolari e sordità neurosensoriale in un modello murino di Norrie malattia. J Neurosci . 2002 ;22 : 4286 -4292.Estratto / LIBERO testo completo
Johnson K, Mintz-Hittner HA, Conley YP, Ferrell RE. X-linked vitreoretinica essudativa causata da una arginina per la sostituzione leucina (R121L) nella malattia di proteine Norrie. Clin Genet . 1996 ; 50 : 113 -115.MedlineWeb of Science
Mintz-Hittner HA, Ferrell RE, Sims KB, et al. Retinopatia periferica nella progenie dei portatori di Norrie mutazioni del gene della malattia. Possibili effetti transplacentare di anormale Norrin. Ophthalmology . 1996 ; 103 :2128 -2134.MedlineWeb of Science
Shastry BS, Hejtmancik JF, Plager DA, Hartzer MK, Trese MT. Linkage e gene candidato analisi di X-linked familiare vitreoretinica essudativa.Genomics . 1995 ; 27 : 341 -344.CrossRefMedlineWeb of Science
Shastry BS, Hejtmancik JF, Trese MT. Identificazione di nuove mutazioni missenso nel gene malattia Norrie associata ad una quattro casi sporadici di familiare vitreoretinica essudativa X-linked e. Hum Mutat . 1997 ; 9 : 396-401.CrossRefMedlineWeb of Science
Torrente I, Mangino M, Gennarelli M, et al. Due nuove mutazioni missense (A105T e C110G) nel gene Norrin in due famiglie italiane con malattia Norrie e familiare vitreoretinica essudativa. Am J Med Genet . 1997 ; 72 : 242 -244.CrossRefMedlineWeb of Science
McDonald NQ, Hendrickson WA. Una superfamiglia strutturale di fattori di crescita che contengono un motivo cistina nodo. cellulare . 1993 ; 73 : 421-424.CrossRefMedlineWeb of Science
. Martoglio B, Dobberstein B. segnale sequenze: più di peptidi solo grassiTrends Cell Biol . 1998 ; 8 : 410 -415.CrossRefMedlineWeb of Science
Fuchs S, Xu SY, Caballero M, et al. Una mutazione puntiforme missense (Leu13Arg) del gene malattia Norrie in un grande cubano affini con Norrie malattia. Hum Mol Genet . 1994 ; 3 : 655 -656.GRATIS Full Text
Yamada K, Limprasert P, Ratanasukon M, et al. Due famiglie tailandesi con malattia Norrie (ND): associazione di due nuove mutazioni missenso con grave fenotipo ND, convulsioni, e un vettore manifesta. Am J Med Genet .2001 ; 100 : 52 -55.CrossRefMedlineWeb of Science
Rivera-Vega MR, Cine-Lopez S, Vaca AL, et al. L’analisi molecolare del gene NDP in due famiglie con Norrie malattia. Acta Ophthalmol Scand . 2005 ; 83 :210 -214.CrossRefMedlineWeb of Science
Shastry BS. Identificazione di una mutazione missense ricorrente nel gene malattia Norrie associato ad un caso simplex di vitreoretinica essudativa.Biochem Biophys Res Commun . 1998 ; 246 : 35 -38. MedlineWeb of Science
Hatsukawa Y, Nakao T, T Yamagishi, Okamoto N, Isashiki Y. nuova mutazione nonsense (Tyr44stop) del gene malattia Norrie in una famiglia giapponese. Br J Ophthalmol . 2002 ; 86 : 1452 -1453.GRATIS Full Text
Allen RC, Russell SR, Streb LM, Alsheikheh A, Pietra EM. Eterogeneità fenotipica associata con una nuova mutazione (Gly112Glu) nella malattia di proteine Norrie. Eye . 2006 ; 20 : 234 -241.CrossRefMedlineWeb of Science
Fuentes JJ, Volpini V, Fernandez-Toral F, Coto E, Estivill X. Individuazione di due nuove mutazioni missense (K58N e R121Q) nella malattia Norrie (ND) gene in due famiglie spagnole. Hum Mol Genet . 1993 ; 2 : 1953 -1955.GRATIS Full Text
Zaremba J, S Feil, Juszko J, et al. La variabilità intrafamiliare del fenotipo oculare in una famiglia polacca con una mutazione missense (A63D) nella malattia gene Norrie. oftalmico Genet . 1998 ; 19 : 157 –MedlineWeb of Science
Chen ZY, Battinelli EM, Woodruff G, et al. Caratterizzazione di una mutazione nel gene NDP in una famiglia con un vettore manifesta femminile.Hum Mol Genet . 1993 ; 2 : 1727 -1729.GRATIS Full Text
Sims KB, Irvine AR, Buona WV. Norrie malattia in una famiglia con un vettore femminile si manifesta. Arch Ophthalmol . 1997 ; 115 : 517 -519.CrossRefMedlineWeb of Science
Shastry BS, Hiraoka M, Trese DC, Trese MT. Sindrome di Norrie e vitreoretinica essudativa in famiglie con femmine portatrici colpite. Eur J Ophthalmol . 1999 ; 9 : 238 -242.MedlineWeb of Science
Puck JM, Willard HF. X inattivazione nelle femmine con malattia X-linked. N Engl J Med . 1998 ; 338 : 325 -328.CrossRefMedlineWeb of Science
Gribnau J, Luikenhuis S, Hochedlinger K, Monkhorst K, Jaenisch R. X scelta cromosoma si verifica indipendentemente dal timing di replicazione asincrona. Biol J cellulare . 2005 ; 168 : 365 -373.
Estratto / LIBERO testo completo
Ross MT, Grafham DV, Coffey AJ, et al. La sequenza di DNA del cromosoma X umano. Nature . 2005 ; 434 : 325 -337.
Articoli citando questo articolo
- Mutazioni recessive in TSPAN12 Causa Retinal displasia grave e familiare essudativa vitreoretinica (FEVR)IOVS 1 mag 2012 53 : 2873 – 2879
- Clinica e molecolare valutazione dei probandi e familiari con Familial essudativa vitreoretinicaIOVS 1 Settembre 2009 50 : 4379 – 4385