SINDROME DI Wildervanck, Sindrome Cervico-Oculo-Acustica, Sindrome (COA).
|
|
Figura 1: paralisi del nervo abducente di sinistra con collo corto e malformato orecchio destro
|
|
|
Figura 2: Klippel-Feil deformità |

Etimologia
Prende il nome da LS Wildervanck un genetista umano olandese che nel 1952, ha descritto il primo caso di quello che ha designato la sindrome cervico-oculo-acustiche (COA). 2
Che cosa è la sindrome Wildervanck?
Questa sindrome, a volte chiamato sindrome cervicooculoacoustica, si compone di tre caratteristiche cliniche principali e diverse minori. I risultati più consistenti sono, sordità percettiva congenita, paralisi abducente paralisi del sesto nervo cranico con Retractio bulbi (sindrome di Duane-Stilling-Turk) con retrazione del globo oculare e restringimento della rima palpebrale su di adduzione ,con problemi di movimento degli occhi, ed, in alcuni casi, deviazione anormale di un occhio rispetto all’altro (strabismo). e una malformazione del collo noto come anomalia di Klippel-Feil(KF). Quest’ultima è caratterizzato da una bassa attaccatura posteriore, collo corto, e movimento del collo limitato secondaria a fusione di più vertebre, si può avere torcicollo e asimmetria facciale. Si può avere Ritardo mentale. Alcuni pazienti possono avere una cisti dermoide epibulbare e ci può essere sovrapposizione con la sindrome di Goldenhar. (RM Winter, M. Baraitser, Londra Dismorfologia Database, Oxford Medical Databases, 2000). La sordità è dovuta alla malformazione delle ossa dell’orecchio medio. Il disturbo movimento degli occhi è conosciuta come sindrome di Duane e si manifesta con l’incapacità di spostare l’occhio (s) lateralmente ed una ritrattazione dei bulbi oculari, quando si muovono verso il naso. La Visione non è interessata. . (RM Winter, M. Baraitser, Londra Dismorfologia Database, Oxford Medical Databases, 2000). La malattia è limitata, o quasi completamente limitata, per le femmine, sollevando la questione della posizione dominante legata al sesso con letalità nel maschio emizigote. Questa sindrome (almeno profonda sordità infantile e di Klippel-Feil malformazione) può essere responsabile di almeno l’1% della sordità tra le donne. La sordità è percettiva ed è stato dimostrato da studi radiologici di essere a causa di una malformazione ossea dell’orecchio Tutte e tre le caratteristiche possono verificarsi in modo indipendente e può anche essere parte di altre sindromi.
Studi dettagliati hanno riscontrato alcune anomalie del midollo spinale e del cervello ma le funzioni di intelligence e motore sono di solito nel range di normalità.
Genetica, Che cosa causa la sindrome Wildervanck?
Non ci è chiaro modello di ereditarietà nella sindrome Wildervanck. Quasi tutti i casi si riscontrano nelle donne che porta alcuni AA a suggerire che una mutazione del gene dominante legato al sesso è responsabile della mortalità nei maschi. Tuttavia, nessun singolo mutazione del gene è stato trovato che porta gli altri a proporre che più geni sono responsabili. Gorlin (1976) Hanno ipotizzato che l’eredità possa essere più facilmente multifattoriale. (RM Winter, M. Baraitser, Londra Dismorfologia Database, Oxford Medical Databases, 2000).Si sospetta che ci siano condizioni poligenici, il che significa che molti fattori genetici possono essere coinvolti. La maggior parte dei casi si verificano sporadicamente. Perché questa sindrome si verifica soprattutto nelle donne, è possibile che questa condizione è ereditarietà dominante X-linked. La mancanza di maschi con sindrome di Wildervanck suggerisce che i maschi affetti hanno caratteristiche più gravi e non sopravvivono a nascita. [2]
2]Wettke-Schafer e Kantner, G. X-linked dominanti ereditati malattie con letalità in emizigoti maschi. Genetica umana. 1983; 64: 1-23.
Kirkham (1969) ha descritto una famiglia che è stata colpita attraverso 5 generazioni con sordità percettiva e in cui 2 membri avevano la sindrome di Duane. Konigsmark e Gorlin (1976) ha favorito l’ereditarietà multifattoriale. Wildervanck (1978) ha dato un esame approfondito del soggetto e ha concluso che è più probabile l’eredità poligenica con limitazione per le femmine. Balci et al. (2002) riportarono un bambino con la sindrome di Wildervanck con le seguenti conclusioni su MRI: diastematomielia del midollo inferiori e midollo cervicale accompagnata da ipoplasia vermiana, ernia tonsillare, e conseguente idrocefalo triventricolare. Gli autori hanno suggerito che i bambini con sindrome di Wildervanck dovrebbero essere indagati per anomalie craniospinale di MRI. Abu-Amero et al. (2014) descrissero un maschio con la sindrome Wildervanck che aveva una microdelezioni nel cromosoma X. Il paziente aveva una sindrome di Duane tipo 1retrazione bilaterale, Klippel-Feil anomalia di tipo 1 che causa una fusione quasi completa dei corpi vertebrali cervicali, e sordità bilaterale con malformazioni dell’orecchio interno che coinvolgono la coclea, vestibolo, e canali semicircolari. La Neuroimaging ha rivelato malformazioni del tronco encefalico inferiore e midollo spinale cervicale con diastematomielia incompleta, come riportato da Balci et al. (2002) . Inoltre, il paziente aveva difetto del setto ventricolare e di un difetto del setto atriale che ha richiesto un intervento chirurgico all’età di 2,5 anni.
Citogenetica
In un paziente di sesso maschile con la sindrome Wildervanck, Abu-Amero et al. (2014) rilevato una cancellazione di circa 3 kb sul cromosoma Xq26.3 (chrX: 137,779,548-137,782,146, NCBI35) utilizzando CGH array. La delezione del gene FGF13 comprendeva ( 300.070 ). La cancellazione non era presente nella madre del paziente o in 1 di 8 fratelli e sorelle affetti. Il padre era morto, ma è stato segnalato per essere stato privo di caratteristiche fenotipiche visto nel figlio. Fare clic qui per vedere i riferimenti in PubMed legate a quelle elencate nel paragrafo precedente.
Vedere anche:
Cremers et al. (1984) ; Everberg et al. (1963) ; Fraser e MacGillivray (1968) ; Kirkham (1969) ; McLay e Maran (1969) ; Strisciuglio et al. (1983) ; Wettke-Schafer e Kantner (1983) ; Wildervanck (1960) ; Wildervanck et al. (1966)
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
METODI
I casi di cui di collo corto con la sordità sono stati avvicinati nel modo seguente: 1. registrazione di una storia dettagliata, altezza e peso percentili e segmento superiore al rapporto di segmento inferiore, 2. esame clinico compreso l’esame degli occhi e fondoscopia 3. tronco cerebrale evocata Risposta uditiva (BERA), raggi X della colonna vertebrale cervicale e ecografia dell’addome. Cariotipo è stato fatto in uno dei pazienti per escludere sindrome di Turner. 2D ECHO è stato fatto su un paziente come un mormorio è stata rilevata all’esame clinico. Valutazione dello sviluppo è stato fatto dallo psicologo clinico. Le principali caratteristiche cliniche dei quattro pazienti sono stati tabulati e studiati.
RISULTATI
I risultati sono riportati nella [Tabella – 1] .
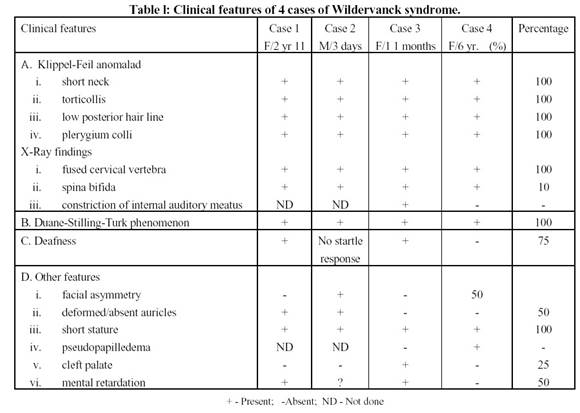
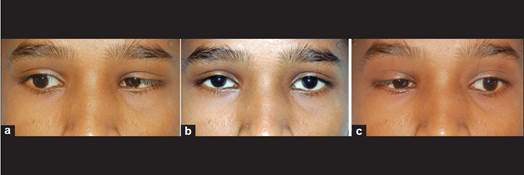
Dei quattro pazienti, due avevano la triade completa, mentre due avevano Klippel-Feil anomala-d con fenomeno di Duane [Figura – 1] .BERA non è stato fatto nel caso 2, in quanto il bambino è stato perso al follow-up. Asimmetria facciale è stato trovato in due pazienti, mentre il ritardo mentale è stato trovato in uno.
DISCUSSIONE
Due risultato insolito presenti nei 4 pazienti studiati in precedenza sono: (1) la presenza di costole cervicali bilaterali, causa 3, e (2) la presenza di associati una malattia cardiaca congenita cioè difetto del setto atriale causa 4. L’associazione delle cardiopatie congenite con sindrome di Klippel-Feil Maggiori dettagli sono stati descritti in letteratura [3] . Nessun precedente relazione circa l’associazione delle cardiopatie congenite con sindrome Wildervanck è stato descritto. Tale constatazione nel nostro caso coincidenza in quanto non è stato descritto in precedenti relazioni o può essere una parte estesa della sindrome.
C’è una transizione graduale nelle caratteristiche cliniche tra forme incomplete e completi della sindrome Wildervanck. Solo un terzo dei pazienti con la Klippel-Feil anomalad hanno ipoacusia e questo può essere puramente conduttiva, neurosensoriale o entrambi [4] ,[5] .
Valutazione radiologica mediante tomografia cranica su uno o entrambi i lati può essere fatto per accertare la presenza di coclea, vestibolo, canali semicircolari e meato uditivo interno [2] . L’esame radiologico delle ossa petrosa non aiuterà nella diagnosi, ma aiuterà considerevolmente nel definire la natura del difetto [2] . Tomografia fatto in due dei quattro pazienti in questa serie non ha rivelato difetti maggiori eccezione costrizione del meato uditivo interno, causa 3. MRI fatto in un caso descritto in letteratura rivelato marcato cerebellare e tronco cerebrale atrofia con invaginazione basilare [6] .
Un Pseudopapilledema era una caratteristica insolita nei due casi di sindrome Wildervanck, recensiti da Kirkham [7] , che porta alla conclusione che l’esame di routine del fondo dovrebbe essere fatto in tutti i casi di sindrome cervico-oculo-acustiche. La presenza di pseudopapilledema è stato pensato per essere una estensione del difetto genetico responsabile della condizione [7] , [8] .
Vi è un consenso circa la modalità di trasmissione della sindrome Wildervanck, ma tutti d’accordo che i fattori genetici sono coinvolti. E’ stata suggerito una trasmissione autosomica dominante a penetranza incompleta ed espressività variabile [8] , [9] . Inoltre, il gene sarebbe in parte limitato dal sesso agisce su uno sfondo poligenica, che viene modificato in base al sesso, rendendo le donne più sensibili rispetto ai maschi all’azione del gene [10] . Un eziologia ambientale, a causa di una sequenza di interruzione vascolare durante lo sviluppo embrionale è stato notato in Klippel-Feil anomalie come Moebius e Polonia sequenze. Una combinazione di difetti (Kiippel-Feil e Moebius) potrebbe indurre il più complesso fenotipo osservata nella sindrome Wildervanck [11] .
Wildervanck ha concluso che eredità poligenica con limitazione per le femmine è più probabile [1] , [8] . [9] . McKusick d’accordo con questa visione [12] . Tre dei quattro pazienti della nostra serie erano femmine. La sindrome è considerato essere letale nei maschi. Il paziente maschio solitario in questa serie è stato osservato a 3 giorni di età e non ha dato seguito successivamente.
La sindrome di Klippel-Feil può sovrapporsi con molte caratteristiche di Turner e di Noonan sindromi, ma la sindrome Wildervanck è l’anomalia congenita multipla più comune in associazione con la sindrome di Duane. Sindrome di Turner è stato indirettamente esclusa in due dei tre pazienti femmine dimostrando ovaie normali in ecografia; mentre cariotipo fatto nella terza era sindrome di Noonan 46XX è stata esclusa in associazione del fenomeno Duane con Klippel-Feil anomalad.
Gli svantaggi nella sindrome rimangono fermi per tutta la vita e apparentemente non influenzano la durata della vita. I pazienti con sordità possono avere bisogno di un apparecchio acustico [1] , [1] .
BIBLIOGRAFIA REFERENCES
|
Wildervanck LS. The cervico-oculo-acousticus syndrome. In: Vinken PJ, Bruyn GW, Myrianthopoulous NC, editors. Congenital Malformations of the Spine and Spinal Cord. Handbook of Clinical Neurology, vol 32. New York: North Holland Publishing Co;1978, pp 123-130. |
|
|
Wilervanck LS, Hoeksema PE, Penning L. Radiological examination of the inner ear of deaf mutes presenting as the cervico-oculo- acoustious syndrome. Acta Otolaryngol 1966; 61:445-453. |
|
|
Masuda H, Arikawa K, Yucia T, Taira A. Total anomalous pulmonary venous connection associated with Klippeil-Feil Syndrome: a case report. Kyobu Geka 1991; 44:417-420. |
|
|
Cremers CWRJ, Hoogland CA, Kuypers W. Hearing loss in cervico-oculo-acousticus syndrome. (Wildervanck syndrome) Arch Otolaryngol 1984; 110:54-57. |
|
|
Dannilidis J, Demetriadis A, Triaridis C. Otological findings in cervico-oculo-auditory dysplasia. J Laryngol Otol 1980; 94:533-544. |
|
|
Hughes PJ, Davies PT, Roche SW, Mathews TD, Lane RJ. Wildervanck or cervico-oculo-acousticus syndrome and MRI findings. J Neurol Neurosurg, Psychiatry 1991; 54:503-504. |
|
|
Kirkhan TH. Cervico-oculo-acousticus syndrome with pseudopapilledema. Arch Dis Child 1969; 44:504-508. |
|
|
Kirkhan TH. Inheritance of Duane’s syndrome. Br J Ophthalmol 1970; 54: 323-329. |
|
|
Konigsmark BW, Gorlin RJ. Genetic hearing loss associated with museuioskeletal abnormalities. In: Genetic and Metabolic Deafness, 1st ed. Philadelphia: WB Saunders; 1976, pp 188-192. |
|
|
Goodman RM, Gorlin RJ. Atlas of the Face in Genetic Disorders, 2nd ed. St Louis: CV Mosby Co; 1977; pp 534-535. |
|
|
Corsello G, Carcione A, Castro IL, GiufWe L. Cervico-oculo- acousticus Wildervanck’s syndrome: a clinical variant of Klippeil-Feil Sequence? Klin Padiatr 1990; 220:176-179. |
|
|
McKusick VA. Mendelian inheritance in man. In: Catalog of Autosomal Dominant, Autosomal Recessive and X Linked Phenotypes, 6th ed. London: John Hopkins University Press; 1983, pp 1105 (Ref no 31460). |
I APPROFONDIMENTO
|
Astratto |
|
Riportiamo un caso di sindrome Wildervanck esporre Klippel-Feil un’anomalia, Duane sindrome di retrazione e sordità congenita. Dal momento che il primo caso è stato segnalato nel 1952, ci sono stati più i rapporti che descrivono questa triade sia completa o incompleta. Il nostro caso ha una triade completa della sindrome con frontale seno ipoplasia. Il nostro caso è unico come la triade è stato associato con ipoplasia del seno frontale, che è un’associazione molto raro.
Parole chiave: Sindrome di Duane, seno frontale ipoplasia, Klippel-Feil Klippel deformità, la sindrome Wildervanck
|
Come citare questo articolo: |
|
Come citare questo URL: |
|
|
|
Nel 1952, Wildervanck, ha descritto una sindrome cervico-oculo-acustica composta da Klippel-Feil deformità, abducente paralisi con globo svincolo (svincolo di Duane), e sordità congenita. [1] L’eziologia non è nota. La maggior parte delle persone sono state donne. I casi con completo così incompleta triade sono state descritte in letteratura. Questo rapporto descrive un caso con ipoplasia seno frontale con triade della sindrome Wildervanck. Questa presentazione è stata raramente descritto in letteratura
|
|
|
Il paziente è un 9-year-old bambina ammessi al AVBR Hospital per la deformità di un orecchio, il collo breve e deviato fin dalla nascita. E ‘nata del matrimonio tra consanguinei. La storia prenatale è stato regolare. Non c’era storia di farmaci o l’esposizione radiologica. Storia di nascita era anche tranquillo senza complicazioni durante il parto e periodo post-parto. Punto di vista evolutivo, era normale. Parametri antropometrici erano suggestivi di bassa statura (altezza – 119 centimetri; <3 ° percentile). All’esame, i suoi segni vitali sono stabili. Orecchio destro era malformato senza visibile canale vestibolare. Orecchio sinistro stava avendo scarico. Il collo è corto con deviazione a destra [Figura 1] . Inoltre, c’era cifoscoliosi delle vertebre toraciche [figura 2] . Il suo occhio destro era più piccola di quanto l’occhio sinistro con una diminuzione involontario intermittente in dimensioni. Gli esami hanno mostrato paralisi sistemica dell’ abducente e del nervo facciale destro. Valutazione oftalmica è stato fatto per piccole dimensioni dell’occhio destro.
|
|
Figura 1: Sinistra abducente paralisi del nervo con collo corto e malformato orecchio destro |

|
|
Figura 2: Klippel-Feil deformità |
Tutti i risultati di cui sopra erano indicativi di sindrome di Wildervanck. Bambino è stato indagato per vari deformità. I suoi rapporti di sangue erano normali. Audiometria rivelato sordità profonda sul lato destro e moderata ipoacusia neurosensoriale sul lato sinistro. Brainstem evocata risposta audiometria è anche indicativo di perdita dell’udito neurosensoriale sul lato sinistro. La tomografia computerizzata (CT) spina dorsale stava mostrando della fusione delle vertebre toraciche, suggestivo di Klippel-Feil deformità. CT seni paranasali sono suggestive del seno frontale ipoplasico [Figura 3] . CT cervello era suggestivo di colesteatoma destra [Figura 4] .Questo era una associazione rara con la sindrome Wildervanck. E’ stato previsto per Il paziente un intervento chirurgico per colesteatoma. Mentre stava anche avendo il collo corto, anestesista ha evidenziato un alto rischio. Quindi, i parenti del paziente si sono rifiutati di optare per un intervento chirurgico; quindi, bambino è stato presentato ad un centro superiore con una migliore gestione competenze per la chirurgia.
|
|
Figura 3: Diritto seno ipoplasia frontale |
|
|
Figura 4: Colesteatoma |
|
|
|
La Sindrome di Wildervanck comprende la triade deformità di Klippel-Feil (fusione di ≥ 1 vertebra cervicale), la sindrome di retrazione di Duane, e la perdita dell’udito. [2] Il collo è corto, fitto, palmato, e immobile. [2] La testa sembra sedersi direttamente sul tronco. Altri deformità vertebrali possono coesistere (spina bifida occulta, Sprengel deformità, e emivertebre, fusione delle costole, costole assenti, cifosi, scoliosi, e impressione basilare). La perdita dell’udito in pazienti con la sindrome Wildervanck può essere neurosensoriale, conduttiva, o mista e può essere accompagnato da malformazioni dell’orecchio esterno, meato esterno acustica, ossicini, e del labirinto osseo. [2] Il nostro caso aveva anche un orecchio malformato destro. L’intelligenza può essere lievemente a gravemente ridotta. [3]
Vi è un consenso circa la modalità di trasmissione della sindrome Wildervanck, ma tutti d’accordo che i fattori genetici sono coinvolti. E’ stata suggerita Trasmissione autosomica dominante a penetranza incompleta ed espressività variabile. [4] , [5] Inoltre, il gene sarebbe in parte il sesso agire limitata su uno sfondo poligenica, che viene modificato in base al sesso, rendendo le donne più sensibili rispetto ai maschi all’azione di il gene. [6] Un eziologia ambientale, a causa di una sequenza di interruzione vascolare durante lo sviluppo embrionale è stato notato in anomalie Klippel-Feil come Moebius e Polonia sequenze. Una combinazione di difetti (Kiippel-Feil e Moebius) potrebbe indurre il più complesso fenotipo osservata nella sindrome Wildervanck. [7]
Wildervanck ha dichiarato che la sordità neurosensoriale dovrebbe essere in tipo; Sono stati riportati anche casi con perdite conduttive o misti. [8] , [9] Solo un terzo dei pazienti con Wildervanck sono stati descritti come aventi perdita dell’udito; anche se, audiometria in nostro paziente ha rivelato un moderato grado di sordità neurosensoriale.
|
|
|
La Sindrome di Wildervanck con ipoplasia del seno frontale e colesteatoma è una associazione molto rara, che non è ancora stato descritto in letteratura. Inoltre, è necessaria l’intervento chirurgico per il drenaggio di colesteatoma. E’ necessario un lavoro di team di esperti per la chirurgia in quanto l’ intubazione può essere difficile a causa della deformità di Klippel-Feil. La possibilità di Wildervanck dovrebbe essere tenuto a mente, mentre la valutazione di un caso di Klippel-Feil deformità.
References
1.Wildervanck LS. A case of Klippel-Feil’s syndrome with abducens paralysis; retraction of the eyeball and deaf-mutism. Ned Tijdschr Geneeskd 1952;96:2752-6. Back to cited text no. 1[PUBMED]
2.Cohen MM, Gorlin RJ. Genetic hearing loss associated with musculoskeletal disorders. In: Gorlin RJ, Toriello JV, Cohen MM, editors. Hereditary Hearing Loss and Its Syndromes. New York, NY: Oxford University Press Inc.; 1995. p. 204-7. Back to cited text no. 2
3.Fraser WI, MacGillivray RC. Cervico-oculo-acoustic dysplasia. (“The syndrome of Wildervanck”). J Ment Defic Res 1968;12:322-9. Back to cited text no. 3[PUBMED]
4.Kirkham TH. Inheritance of Duane’s syndrome. Br J Ophthalmol 1970;54:323-9. Back to cited text no. 4
[PUBMED]
5.Konigsmark BW, Gorlin RJ. Genetic hearing loss associated with museuioskeletal abnormalities. In: Genetic and Metabolic Deafness. 1 st ed. Philadelphia: WB Saunders; 1976. p. 188-92. Back to cited text no. 5
6.Goodman RM, Gorlin RJ. Atlas of the Face in Genetic Disorders. 2 nd ed. St. Louis: CV Mosby Co.; 1977. p. 534-5. Back to cited text no. 6
7.Corsello G, Carcione A, Castro L, Giuffrè L. Cervico-oculo-acusticus (Wildervanck’s) syndrome: A clinical variant of Klippel-Feil sequence? Klin Padiatr 1990;202:176-9. Back to cited text no. 7
8.Wildervanck LS. The cervco-oculo-acoustic syndrome. Handbook of Clinical Neurology. Vol. 32. New York: North Holland Publishing Co.; 1978. p. 123-30. Back to cited text no. 8
9.Oe K, Mori K, Konno T, Yoneda T, Ueyama K, Yamagishi M. Ruptured aneurysm of the sinus of Valsalva with Wildervanck syndrome (cervico-oculo-acoustic syndrome), blepharoptosis and short stature: Case report. Circ J 2007;71:1485-7. Back to cited text no. 9
Importante
E ‘possibile che il titolo principale della relazione Sindrome Wildervanck non è il nome che vi aspettavate. Si prega di controllare isinonimi dell’annuncio per trovare il nome alternativo (s) e il disordine suddivisione (s) oggetto della presente relazione.
- Sindrome di cervico-oculo-Acoustic
- Sindrome COA
- Nessuno
Sindrome Wildervanck, nota anche come sindrome cervicooculoacoustica, è una malattia genetica rara che colpisce soprattutto le donne. La malattia è caratterizzata da una condizione scheletrica nota come sindrome di Klippel-Feil (KFS); anomalie idell’occhio (oculari) di alcuni movimenti (ad esempio, la sindrome di Duane ); e / o la presenzae alla nascita di una ipoacusia(congenita) .
Nei soggetti con KFS, c’è unione anormale o fusione di due o più ossa della colonna vertebrale (vertebre) all’interno del collo (vertebre cervicali). La Sindrome di Duane è caratterizzata da limitazione o l’assenza di certi movimenti oculariorizzontali ; retrazione o “tirarsi indietro” del bulbo oculare nella occhio cavità (orbita) durante il tentativo di guardarsi dentro; e, in alcuni casi, deviazione anormale di un occhio rispetto all’altro (strabismo). In alcuni individui affetti, ulteriori anomalie fisiche possono anche essere presenti. Nella maggior parte dei casi, la sindrome Wildervanck sembra verificarsi in modo casuale per ragioni sconosciute (sporadicamente).
Bibliografia
Imaizumi,-K
[Wildervanck (cervico-oculo-acoustic) syndrome]
Ryoikibetsu-Shokogun-Shirizu. 2001; (33): 361-2
Hayashi,-M
[Wildervanck syndrome, cervico-oculo-acoustic syndrome]
Ryoikibetsu-Shokogun-Shirizu. 2000; (30 Pt 5): 260-2
Haciyakupoglu,-G; Pelit,-A-A; Altunbasak,-S; Soyupak,-S; Ozer,-C
Crocodile tears and Dandy-Walker syndrome in cervico-oculo-acoustic syndrome.
J-Pediatr-Ophthalmol-Strabismus. 1999 Sep-Oct; 36(5): 301-3
Brodsky,-M-C; Fray,-K-J
Brainstem hypoplasia in the Wildervanck (cervico-oculo-acoustic) syndrome.
Arch-Ophthalmol. 1998 Mar; 116(3): 383-5
Wang,-Y; Shi,-X; Wang,-Z
[Wildervanck or cervico-oculo-acoustic syndrome]
Lin-Chuang-Er-Bi-Yan-Hou-Ke-Za-Zhi. 1997 Nov; 11(11): 499-501
Kumar,-A; Chaudhary,-D; Gupta,-S-K
Wildervanck syndrome.
Australas-Radiol. 1996 May; 40(2): 160-1
Stoll,-C; Alembik,-Y; Dott,-B
Association of Duane anomaly with mental retardation, cardiac and urinary tract abnormalities: a new autosomal recessive condition?
Ann-Genet. 1994; 37(4): 207-9
Kose,-G; Ozkan,-H; Ozdamar,-F; Kavukcu,-S; Ozaksoy,-D
Cholelithiasis in cervico-oculo-acoustic (Wildervanck’s) syndrome.
Acta-Paediatr. 1993 Oct; 82(10): 890-1
Gupte,-G; Mahajan,-P; Shreenivas,-V-K; Kher,-A; Bharucha,-B-A
Wildervanck syndrome (cervico-oculo-acoustic syndrome).
J-Postgrad-Med. 1992 Oct-Dec; 38(4): 180-2
Sholapurkar,-S-L; Dhall,-G-I
Cervico-oculo-acousticus syndrome with pregnancy.
Gynecol-Obstet-Invest. 1991; 31(2): 116-8
Hughes,-P-J; Davies,-P-T; Roche,-S-W; Matthews,-T-D; Lane,-R-J
Wildervanck or cervico-oculo-acoustic syndrome and MRI findings.
J-Neurol-Neurosurg-Psychiatry. 1991 Jun; 54(6): 503-4
Corsello,-G; Carcione,-A; Castro,-L; Giuffre,-L
Cervico-oculo-acusticus (Wildervanck’s) syndrome: a clinical variant of Klippel-Feil sequence?
Klin-Padiatr. 1990 May-Jun; 202(3): 176-9
West,-P-D; Gholkar,-A; Ramsden,-R-T
Wildervanck’s syndrome–unilateral Mondini dysplasia identified by computed tomography.
J-Laryngol-Otol. 1989 Apr; 103(4): 408-11
Cohen,-M-M Jr; Rollnick,-B-R; Kaye,-C-I
Oculoauriculovertebral spectrum: an updated critique.
Cleft-Palate-J. 1989 Oct; 26(4): 276-86