TRASMISSIONE “X LINKED”
Sordità Legate Al Cromosoma XTrasmissione ereditaria recessiva legata al sesso “X-Linked”
Indice
Cos’è l’ereditarietà recessiva legata all’X?
Ereditarietà dominante legata all’X
Analisi del portatore e analisi in gravidanza
Elenco di ipoacusie a Trasmissione “X-Iinked”
Ipoacusia Autosomica recessiva (AR)
Ipoacusia Autosomica dominante (AD)
Ipoacusia a Trasmissione mitocondriale
LE IPOACUSIE CONGENITE
Mentre fino a qualche anno fasi riteneva che le cause genetiche rappresentassero un terzo dei casi di ipoacusia nel bambino, attualmente grazie allo sviluppo delle diagnosi molecolari è stato possibile ricollegare a una causa genetica la maggioranza dei casi sporadici di ipoacusia, in precedenza classificati come “causa ignota”12.
Oggi pertanto si stima che circa il 75% delle ipoacusie congenite sia riferibile a cause genetiche (sindromiche nel 30%) e che la restante parte sia da ricondurre a cause ambientali, soprattutto infettive.
Le Ipoacusie genetiche
Le ipoacusie genetiche sono, nella maggior parte dei casi, malattie monogeniche ossia dovute alla lesione di un singolo gene che causa generalmente una lesione Cocleare.
La lesione è rappresentata da mutazioni della sequenza di base del DNA con modalità di alterazione differenti (sostituzione/inserzione/delezione).
Le modalità di trasmissione di una ipoacusia genetica sono:
Sordità Legate Al Cromosoma X Trasmissione ereditaria recessiva legata al sesso “X-Linked” Legata al cromosoma X (1%)
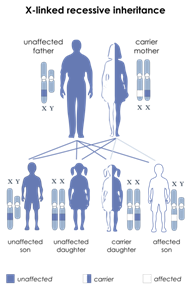
Rappresentano il 2-3% delle forme non sindromiche. Data la localizzazione della mutazione sul cromosoma X, la malattia colpisce generalmente solo i maschi i quali, possedendo un solo cromosoma X, sono definiti emizigoti per la mutazione. Le loro madri, avendo un cromosoma X normale ed uno con la mutazione, sono definite portatrici sane della malattia. Le donne portatrici sane trasmettono la malattia al 50% dei figli maschi.
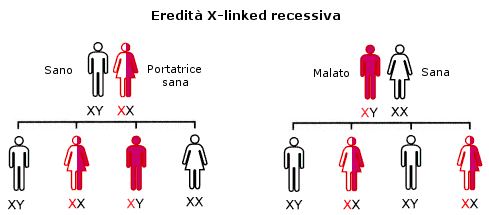
Una malattia monogenica si dice a trasmissione ereditaria recessiva legata al sesso quando il gene coinvolto è localizzato su un cromosoma sessuale (X o Y); se il gene si trova sul cromosoma X si parla di trasmissione ereditaria legata all’X (X-linked) e il carattere patologico si manifesta di solito solo in individui di sesso maschile in quanto portano il solo cromosoma X con l’allele mutato (emizigoti); esempi di malattie recessive legate all’X sono la distrofia muscolare di Duchenne, sindrome dell’X-fragile, emofilia A e B. Un individuo di sesso femminile avendo una sola copia alterata di un gene (eterozigote) generalmente non manifesta la malattia ed è indicato come “portatore sano”. Nella Fig.1 è rappresentata la situazione più frequente, cioè l’incontro di una femmina “portatrice sana” e un maschio normale; per un dato gene del cromosoma X l’allele normale è indicato con X, quello mutato con x. Una madre eterozigote (Xx) per una mutazione produce ovuli che presentano l’allele nomale X o l’allele mutato x. Il padre da origine a spermatozoi con l’allele X o con il cromosoma Y. Dall’incontro di una femmina “portatrice sana” (eterozigote) e un maschio normale, a seconda del tipo di gameti che si fondono al momento della fecondazione si verifica che:
· in media il 50% dei figli maschi risulterà affetto (xY) e potrà trasmettere l’allele mutato x a tutte le sue eventuali figlie
· in media il 50% delle figlie di madri eterozigoti, sarà eterozigote e avrà una probabilità del 50% di trasmettere il cromosoma X con l’allele mutato (x) ai figli.
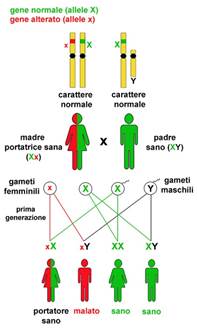
Fig.1- Trasmissione ereditaria di una malattia genetica legata al cromosoma X (X-linked)
Le malattie X-linked raramente possono colpire anche le donne. Una patologia può manifestarsi:
· in figlie (omozigoti xx) di madre eterozigote per la mutazione (Xx) e padre affetto (xY), in quanto ereditano l’allele mutato x da entrambi i genitori;
· in femmine eterozigoti (Xx) in cui si è inattivato preferenzialmente il cromosoma X con l’allele normale;
· in femmine con monosomia del cromosoma X (sindrome di Turner) in cui è presente solo il cromosoma X con la mutazione.
Con questa modalità il gene responsabile della sordità è situato sul cromosoma X, anziché su uno dei cromosomi autosomici.La sordità si manifesta nei maschi, che sono dotati di un solo cromosoma X (fig. 35c).Si ritiene che ‘1-2% delle sordità genetiche siano trasmesse con questa modalità, X-linked o legata al sesso, sono disturbi ereditati attraverso i geni sul cromosoma X. Il gene in causa è situato sul cromosoma X. I maschi che posseggono solo un cromosoma X sono più sensibili ai sintomi clinici ipoacusia di una malattia X-linked, questi trasmetteranno l’X portatore della mutazione genetica alle loro figlie che però saranno udenti. Nelle donne, infatti, l’X portatore della mutazione genetica è “compensato” dal secondo X normale (ipoacusia recessiva legata all’X).
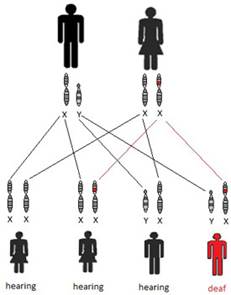
Raramente, come nella sindrome di Alport, l’ipoacusia è dominante legata al cro- mosoma X e le femmine sono allora anche colpite, ma in modo meno grave dei maschi. Figura 2 mostra un esempio in cui la madre è un vettore inalterata e il padre ha una copia normale del gene. Il figlio colpito ha ereditato il cromosoma X anomalo dalla madre. Nei disturbi X-linked, i padri affetti di solito passano il gene anomalo per le loro figlie, ma non per i loro figli. Più del 80% dei pazienti con sindrome di Alport ha una malattia legata al cromosoma X associata a perdita dell’udito neurosensoriale, malattie renali e anomalie oculari (Kruegel, Rubel, e Gross, 2013; Rheault, 2012).
|
|
|
|
|
|
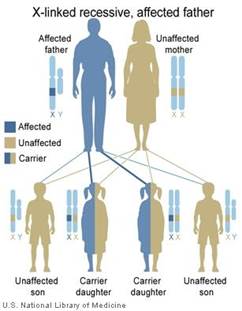
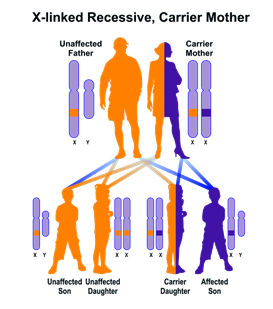
Fig. 2. ereditarietà X-linked
Legata al cromosoma Y (<1%)
Il gene in causa è situato sul cromosoma Y e pertanto interessa esclusivamente il sesso maschile. Non sono state riscontrate altre anomalie associate. Gli unici casi riscontrati appartengono ad una famiglia cinese con alterazione del gene POU3F4’.
Cos’è l’ereditarietà recessiva legata all’X?
Il cromosoma X ha molti geni che sono importanti per la crescita e lo sviluppo. Il cromosoma Y è più piccolo e ha meno geni. Le femmine hanno 2 cromosomi X (XX) e quindi se uno dei geni su un cromosoma X ha una mutazione, il gene normale sull’altro cromosoma X può compensare la copia mutata. Se questo accade la femmina è portatrice sana della malattia legata all’X. Essere portatori significa che non si ha la malattia ma si porta una copia del gene mutato. In alcuni casi le femmine mostrano segni leggeri della malattia. I maschi hanno un cromosoma X e un Y (XY) e quindi se uno dei geni su un cromosoma X del maschio ha una mutazione, non c’è un’altra copia di quel gene per compensare la copia mutata. Questo significa che sarà affetto dalla malattia, che quando viene ereditata in questo modo viene chiamata malattia recessiva legate all’X. Alcuni esempi di malattie legate all’X includono la Sindrome di Norrie , la Sindrome di Alport,sindrome oto-palato-digitale (OPD) l’emofilia e la distrofia muscolare Duchenne.
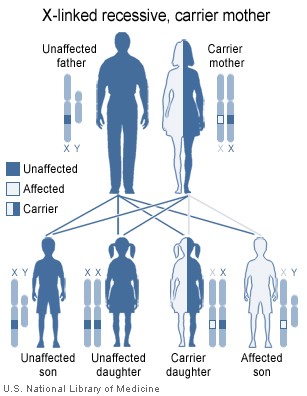
Schema di trasmissione genetica con gene X recessivo da madre portatrice sana
Fig.3: Come vengono trasmesse le malattie legate all’X dalle femmine portatrici
Nel caso si tratti di anomalie recessive , la malattia colpisce praticamente solo i maschi nati da madri portatrici (clinicamente sane).
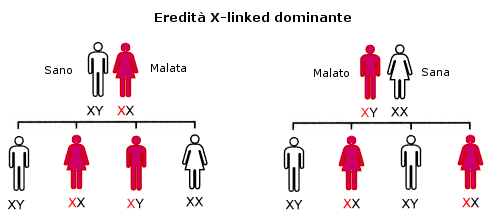
Ereditarietà dominante legata all’X
Sebbene la maggior parte di malattie legate al cromosoma X segua una trasmissione recessiva, vi sono rare forme di malattie genetiche legate al cromosoma X che seguono una trasmissione dominante. Questo vuol dire che anche se una femmina eredita un cromosoma X normale ed un cromosoma X con una mutazione, il gene mutato ha il sopravvento sul gene normale e quindi si manifesta la malatia. Se un maschio eredita il cromosoma X con la mutazione sarà affetto perchè i maschi hanno un solo cromosoma X. Una femmina ha quindi 1 probabilità su 2 (50%) di trasmettere la malattia sia ai figli maschi che alle femmine. Un maschio affetto avrà tutte le figlie femmine affette ma tutti i maschi normali (perchè a questi trasmetterà solo il cromosoma Y).
Le malattie X-linked dominanti sono rare. In questo caso una madre affetta ha un 50% di probabilità d trasmettere la malattia ai propri figli (maschi o femmine che siano), mentre un padre affetto trasmette la malattia solo alle figlie.
Punti da ricordare
· Le femmine portatrici hanno il 50% di possibilità di passare il gene mutato. Se un figlio eredita un gene mutato da sua madre, allora sarà affetto dalla malattia. Se una figlia eredita un gene mutato sarà portatrice come sua madre.
· Un maschio che ha la malattia legata all’X passerà sempre il gene mutato a sua figlia che sarà quindi una portatrice. Un maschio non passerà mai un gene mutato a suo figlio.
· Un gene mutato è presente per tutta la vita e raramente può essere corretto.
· Un gene mutato non è qualcosa che può essere preso da altre persone. Quindi si può anche essere donatore di sangue, per esempio (ad eccezione di soggetti con malattie genetiche del sangue).
· Le persone spesso si sentono colpevoli a causa di una malattia genetica che si trova in famiglia. È importante ricordare che non è colpa di nessuno e nessuno ha fatto nulla per causarne la comparsa.
Se una femmina portatrice ha una figlia, passerà o il cromosoma X con il gene normale, o il cromosoma X con il gene mutato. Ogni figlia quindi 1 probabilità su 2 (il 50%) di ereditare il gene mutato. Se questo accade la figlia sarà portatrice come la madre. C’è anche 1 probabilità su 2 (il 50%) che la figlia erediti il gene normale. Se questo accade sarà totalmente sana rispetto la malattia. Questo rischio rimane lo stesso per ogni figlia.
Se un maschio che ha la malattia legata all’X ha una figlia le trasmetterà sempre il gene mutato. Questo perché i maschi hanno solamente un cromosoma X e passano sempre questo alle loro figlie. Tutte le sue figlie saranno portatrici. Le figlie di solito non avranno la malattia ma sono a rischio di avere figli affetti.
Se un maschio che ha una malattia legata all’X ha un figlio, suo figlio non erediterà mai il gene mutato sul cromosoma X. Questo perché i maschi sempre passano il loro cromosoma Y ai loro figli ( se passano il loro cromosoma X avranno una figlia).
Cosa accade se un bambino é la prima persona nella famiglia ad avere la malattia?
Qualche volta un bambino nato con una malattia genetica legata all’X può essere la prima persona ad essere affetta nella famiglia. Questo può accadere perché una nuova mutazione genetica é avvenuta per la prima volta o nell’uovo o nello spermatozoo che originarono il bambino. Quando questo accade, nessuno dei genitori del bambino é portatore. Comunque il bambino affetto che ora ha il gene mutato, può passarlo ai suoi bambini.
Analisi del portatore e analisi in gravidanza
Diverse opzioni possono essere disponibili per le persone che hanno una storia familiare di malattia genetica legata all’X. L’analisi del portatore può essere disponibile per le femmine per vedere se sono portatrici del gene mutato. Questa informazione può essere utile quando si pianificano gravidanze. Per alcune malattie legate 8 all’X é possibile fare l’analisi in gravidanza per vedere se il bambino ha ereditato la malattia (ulteriori informazioni circa queste analisi sono disponibili negli opuscoli CV e amniocentesi). In questi casi si consiglia di discuterne con lo specialista in Genetica Medica (consulenza genetica).
Situazioni riguardanti altri membri familiari Se qualcuno in famiglia ha una malattia legata all’X o é portatore, potrebbe essere utile discutere con altri membri della famiglia. In questo modo si offre ad altre femmine membri della famiglia l’opportunità di fare un’analisi del sangue per vedere se sono portatrici, se lo desiderano. Queste informazioni possono essere utili ad aiutare altri membri della famiglia a fare una diagnosi. Questo potrebbe essere particolarmente importante per i membri della famiglia che hanno già figli o che vorrebbero avere figli in futuro. Alcune persone trovano difficile di parlare della malattia genetica ad altri membri della famiglia. Essi possono preoccuparsi di causare ansia nella famiglia. In alcune famiglie le persone hanno perso il contatto con i parenti e può essere difficile contattarli. Lo specialista in genetica ha molta esperienza con le famiglie in queste situazioni ed è in grado di offrire un aiuto per discutere la situazione con altri membri della famiglia
Elenco di ipoacusie a Trasmissione “X-Iinked” Legata al cromosoma X
La sindrome di Alport è una condizione genetica caratterizzata dalla progressiva perdita di funzione renale e uditiva. La sindrome di Alport può inoltre interessare gli occhi. La presenza di sangue nelle urine (ematuria) è quasi sempre riscontrabile nella patologia.Nella maggior parte (80-85%) dei pazienti affetti da Sindrome di Alport, la patologia è trasmessa con un’ereditabilità legata all’X, dovuta a mutazioni nel gene COL4A5. Una condizione si considera legata all’X se il gene coinvolto nella malattia è localizzato sul cromosoma X. Nei soggetti di sesso maschile, che possiedono un solo cromosoma X, una copia alterata del gene COL4A5 è sufficiente per causare la Sindrome di Alport in forma severa, fino ad arrivare all’insufficienza renale. Nelle femmine, che possiedono due cromosomi X, la mutazione di una copia di COL4A5 solitamente provoca ematuria, ma nella maggior parte dei casi non arrivano mai all’insufficienza renale. Una interessante caratteristica delle malattia a trasmissione legata all’X è che il padre non le trasmette mai ai figli maschi.
La sindrome di Norrie (o malattia di Norrie, o displasia oculo-acustico-cerebrale), è una malattia genetica molto rara, legata al generecessivo NDP presente sul cromosoma X, che causa cecità, sordità e ritardo mentale. Colpisce solo i maschi, mentre le femmine sono portatrici sane.
La sindrome oto-palato-digitale (OPD) è una malattia genetica rara, che associa displasia scheletrica, ipoacusia, palatoschisi e viso caratteristico (con ipertelorismo, radice nasale allargata, bozze frontali prominenti, naso piccolo piatto e rime palpebrali rivolte in basso e verso l’esterno). La patologia riconosce un’eziologia genetica ed una modalità di trasmissione di tipo X-linked dominante (tipo I) o recessiva (tipo II). Il gene della forma tipo II responsabile è stato mappato al locus Xq28.
La sindrome di Wildervanck, nota anche come sindrome cervicooculoacoustica, è una malattia genetica rara che colpisce soprattutto le donne. La malattia è caratterizzata da sordità congenita percettiva, una condizione scheletrica nota come sindrome di Klippel-Feil (KFS); anomalie di alcuni movimenti dell’occhio con Retractio bulbi (ad esempio, la sindrome di Duane); Nella maggior parte dei casi, la sindrome Wildervanck sembra verificarsi in modo casuale per ragioni sconosciute (sporadicamente). Poiché la malattia colpisce soprattutto le donne, alcuni ricercatori suggeriscono che la sindrome Wildervanck può essere trasmessa come carattere dominante X-linked.
altre ipoacusie X-Linked sono ad esampio:DFNX1 (DFN2),DFNX2 (DFN3), DFNX4 (DFN6)
DFNX (per Deafness X Linked) (per Deafness Autosomal Dominant), DFNB (per Deafness Autosomal Recessive), Si attribuisce poi un numero per ordine di scoperta (Tabelle I—IV).
Tabella I – Lista dei loci e dei geni delle ipoacusie X-Linked (da Van Camp e Smith 2010)
|
Locus |
Posizione |
Gene |
Referenze
|
|
DFNXI |
Xq22 |
PRPSI |
Liuetal.. 2010
|
|
DFNX2 |
Xg21.1 |
POU3F4 |
DeKoketal., 1995
|
|
DFNX3 |
Xp21.2 |
sconosciuto |
Lalwani et al., 1994 |
|
DFNX4 |
Xp22 |
sconosciuto |
del Castillo et aI.. 1996
|
|
DFNX5 |
Xq23-q27.3 |
sconosciuto |
Wang et al , 2006 |
|
Locus (OMIM) |
Gene (OMIM) |
Riferimento |
|
DFNX1 (DFN2) |
||
|
DFNX2 (DFN3) |
||
|
DFNX4 (DFN6) |
||
Ipoacusia Autosomica recessiva (AR) (76%)
E’ la modalità più frequente. infatti si stima che circa tre quarti delle ipoacusie non sindromiche si trasmettano in modalità AR.Quando una malattia si trasmette in ipoacusia a trasmissione autosomica recessiva della moda, due copie del gene anomalo causano la malattia. Gli individui che ereditano un solo gene anomalo e un gene normale sono indicati come vettori e non sono colpiti dalla malattia. I genitori portatori di un solo allele anomalo del gene in causa (portatori eterozigoti) Sono normoudenti e spesso sono inconsapevoli del loro stato di portatore fino a quando non hanno un figlio affetto. Gli individui affetti da una malattia autosomica recessiva di solito sono il risultato di accoppiamenti tra due vettori. La Fig.1 illustra il potenziale risultati di tale accoppiamento, in cui il 25% della prole sono affetti da disturbo in questione, il 50% sono portatori sani, e il 25% sono liberi della malattia e con il gene anormale. La Consanguineità , o la presenza di un comune antenato tra la coppia, aumenta il rischio di una malattia autosomica recessiva. La Sindrome di Usher mostra un modello di ipoacusia autosomica recessiva. La prevalenza della sindrome di Usher è di 3 a 4 soggetti affetti si 100.000 nati (Ahmed, Riazuddin, Riazuddin, & Wilcox, 2003), con tassi di prevalenza più elevati in alcune popolazioni, tra cui la popolazione Acadian in Louisiana (Ouyang et al., 2003) e la popolazione ebrea Ashkenazi (Guha et al., 2012). Il tipo più grave di sindrome di Usher è caratterizzata da una sordità profonda neurosensoriale congenita e da progressiva perdita della vista (Liu et al., 2013). Garantire una diagnosi precoce della sindrome di Usher può aiutare i pazienti a prepararsi per il loro futuro. La sindrome di Usher può rappresentare la metà di tutti i casi di concomitante sordità / cecità (Yan e Liu, 2010).
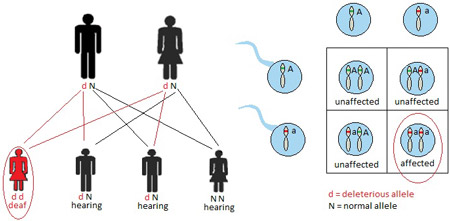
Fig.1 ereditarietà autosomica recessiva

Ipoacusia Autosomica dominante (AD) (22%)
In un modello di ereditarietà autosomica dominante , un bambino eredita una copia normale di un gene da un genitore e un gene anomalo dall’altro genitore. Nelle famiglie affette da sordità autosomica dominante, uno dei genitori è sordo e porta su un singolo allele del gene la mutazione patogena, che trasmetterà a metà dei suoi figli, che quindi saranno sordi. Il gene anomalo domina il gene normale, così una copia di un gene anormale è sufficiente a causare una malattia autosomica dominante (Fig. 2).La mutazione dell’allele interessato può. per esempio. produrre una proteina anormale, che impedisce la funzione della proteina normale prodotta dall’allele sano. La Sindrome di Waardenburg è la causa più comune di perdita di udito sindromica autosomica dominante, che colpisce 1 su 42.000 persone (Read & Newton, 1997). Quando un genitore ha la sindrome di Waardenburg, ogni bambino di quel genitore ha una probabilità del 50% di essere affetti da sindrome di Waardenburg, assumendo l’altro genitore non ha la sindrome. Il restante 50% della prole in questo accoppiamento sarà inalterato. Caratteristiche tipiche della sindrome di Waardenburg includono perdita dell’udito neurosensoriale, un ciuffo bianco, occhi celesti o di colore diverso, e gli occhi distanziati. Una grande quantità di espressività variabile esiste in Waardenburg sindrome, in modo che i pazienti possono avere una qualsiasi combinazione di caratteristiche, tra cui un udito normale (de Sousa Andrade et al., 2012). Waardenburg sindrome rappresenta circa il 2% dei casi di sordità profonda congenita (de Sousa Andrade et al., 2012).Va notato che, in rari casi, alcuni disturbi autosomica dominante verificarsi a causa di una nuova mutazione nel bambino e nessuno dei genitori ha il disturbo. L’espressività è però variabile in questa modalità di trasmissione: la gravità della ipoacusia può essere variabile tra i vari soggetti colpiti.
Quando l’ipoacusia è sindromica i segni associati alla sindrome possono essere assenti o lievi in alcuni sordi della famiglia e alcuni membri portatori dell’ alterazione genetica possono essere udenti.
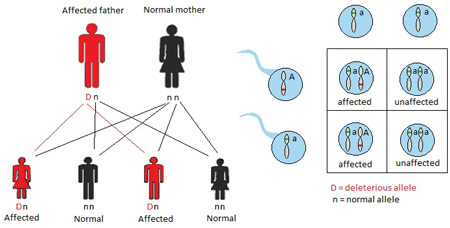

Fig.2 ereditarietà autosomica dominante
Ipoacusia a Trasmissione mitocondriale (1%)
Il genoma mitocondriale è un frammento di DNA situato nel mitocondrio. La trasmissione dell’anomalia mitocondriale è esclusivamente matrilineare. Quando un gene dell’ipoacusia è situato sul DNA mitocondriale. uomini e donne possono essere sordi, ma solamente le donne potranno trasmettere l’ipoacusia ai loro figli che saranno in teoria tutti sordi.
Sebbene mutazioni mitocondriali geni rappresentano una piccola percentuale di casi di perdita di udito non sindromica, sono importante notare per la loro modalità di trasmissione e la loro associazione con una insolitamente alta suscettibilità alla ototossicità da aminoglicosidici (Chen, egli, Fu, e Dong, 2011). I mitocondri sono organelli cellulari responsabili della produzione di energia. Si trovano al di fuori del nucleo e hanno il loro DNA e geni, separati dai geni nucleari. I mitocondri sono presenti in cellule uovo, ma non in cellule spermatiche, quindi sono ereditate esclusivamente attraverso la linea materna. Una donna con una mutazione mitocondriale passerà la mutazione a tutti i suoi figli, mentre un uomo con una mutazione mitocondriale non dia mai a nessuno dei suoi figli. Eredità mitocondriale è dimostrato in Figura 4. Le mutazioni nel mitocondriale MT-RNR1 gene causa un aumento della suscettibilità alle ototossicità aminoglicoside indotta, a volte causando sordità profonda dopo una sola dose di antibiotici aminoglicosidici (Van Camp & Smith, 2000). Lo screening neonatale per le mutazioni mitocondriali che portano a ototossicità aminoglicoside indotta non è ancora disponibile. Il test genetico dovrebbe essere richiesto, se un modello di ereditarietà materna è osservato (Figura 4) e il bambino è influenzata dalla somministrazione di antibiotici aminoglicosidici. I test genetici di una mutazione mitocondriale può identificare altri membri della famiglia o future gravidanze a rischio di insorgenza precoce perdita sostanziale dell’udito, quindi consentire l’adozione di misure preventive. In questi casi, i genitori si consiglia di evitare la somministrazione di antibiotici aminoglicosidi e cercare trattamenti antibiotici alternativi.
Figura 4. ereditarietà mitocondriale
L’ipoacusia è spesso severa e si manifesta dopo esposizione a antibiotici aminoglicosidici. Alcune fonne sono sindromiche e coinvolgono in particolare il sistema nervoso:
MELAS: Mitochondrial Encephalopathy Lactic Acidosis Stroke-like episodes
KSS: Kearns — Sayre Syndrome
MERRF: Myoclonic Epilepsy and Ragged Red Fibers
MDD: Maternal Inherited Diabetes and Deafness.
Più di 60 geni implicati nella perdita di udito non sindromica sono stati identificati (Van Camp & Smith, 2013). Circa l’80% dei casi di perdita di udito non sindromica genetiche sono ereditate in modo autosomico recessivo, mentre quasi il 20% dimostra una modalità autosomica dominante di eredità (Hilgert, Smith, e Van Camp, 2009). X-linked e geni mitocondriali rappresentano una piccola percentuale di casi (Hilgert et al., 2009). Il gene più comune che causa la perdita di udito non sindromica è il GJB2gene, noto anche come Connexin 26 o DFNB1 (Angeli, Lin, e Liu, 2012). I conti gene connessina 26 per circa la metà di tutte le cause autosomica recessiva di sordità sindromica (Kenneson, Van Naarden Braun, e Boyle, 2002). La maggior parte di questi pazienti sono nati da genitori normali, udito, che sono entrambi portatori di mutazioni nello stesso gene (Angeli et al., 2012).
Loci e geni coinvolti nelle ipoacusie non sindromiche
L’analisi molecolare in famiglie con casi di ipoacusia genetica ha permesso di definire il locus ossia le regioni del genoma contenenti il gene interessato. Il gene spesso è stato identificato in un secondo tempo e in qualche caso un locus ha dimostrato di contenere i due geni dell’ipoacusia. È stata fissata una codifica internazionale pci denominare ogni locus delle ipoacusie non sindromiche. Il codice inizia con DFNA
Tabella II a – Lista dei loci e dei geni delle ipoacusie non sindromiche dominanti
(daVan Camp e Smith 2010
|
Locus |
Posizione |
Gene Referenze |
|
DFNAI |
5q31 |
DIAPHI Léon et al, 1992 |
|
Lynchetal., 1997 |
||
|
DFNA2A |
lp34 |
KCNQ4 Coucke et al., 1994 |
|
DFNA2B |
lp35 I |
GJB3(in Xiaetal., 1999 passato CX3I) |
|
DFNA3A |
13q1 1-q12 |
GJB2(in Chaib et al., 1994 |
|
DFNA3B |
13q12 |
GJB6(in Grifaetal,. 1999 passato CX3O) |
|
DFNA4 |
19q13 |
MYH14 Chen et al., 1995, Donaudy et al, 2004 |
|
DFNA5 |
7p15 |
DFNA5 Van Camp et al, 1995 |
|
DFNA6 |
4pl6.3 |
WFSI Lesperance et al., 1995: Van Camp et al., 1999; |
|
DFNA7 |
1q21-q23 |
sconosciuto Fagerheim et al.. 1996 |
|
DFNA8 |
vedi DFNAI2 |
|
|
DFNA9 |
14ql2-q13 |
COCH Manolis et al.. 1996 |
|
DFNÀIO |
6q22-q23 |
EYA4 ONeili et aI. 1996 |
|
Wayne et al.. 2001 |
||
|
DFNAI I |
1 lql2 3-q2l |
MYO7A Tamagawa et al.. 1996 |
|
DFNA12 |
I Iq2224 |
TECTA Verhoeven et aI.. 1997 |
|
Verhoeven et al,. 1998 |
||
|
DFNAI3 |
6p2l |
COLI 1A2 Brown et al. 1997 |
|
McGuirt et al.. 1999 |
||
|
DFNAI4 |
vedi DFNA6 |
|
|
DFNAI5 |
5g3I |
POIJ4F3 Vahava et al.. 1998 |
|
DFNAI6 |
2q24 |
sconosciuto Fukushima et al., 1999 |
|
DFNAI7 |
22q |
MYH9 Lalwaniet al. 1999 |
|
DFNAI8 |
3q22 |
Sconosciuto Bonsch et al.. 200! |
|
DFNAI9 |
10 (pericentr.) |
sconosciuto The Molecular Biology of Hearing and Deafness, Bethesda, October 8l I. 1998 (Green et al., abstract 107) |
|
DFNA2O |
17q25 |
ACTG1 Moreli et al .2000. Yang et al,. 2000, Zhu et aL. 2003. van |
|
DFNA2I |
6p21 |
sconosciuto Kunst et al.. 2000 |
|
DFNA22 |
6gl3 |
MYO6 Melchionda et al.. 2001 |
|
DFNA23 |
I4q2l-q22 |
sconosciuto Salam et al.. 2000 |
|
DFNÀ24 |
4q |
sconosciuto Hafner et al.. 2000 |
|
DFNA25 |
12g21-24 |
sconosciuto Greene et aI.. 1999 |
|
DFNA26 |
vedi DFNA2O |
|
|
DFNA27 |
4q12 |
sconosciuto Frideil et aI.. 999 |
|
DFNA2S |
8q22 |
TFCP2L3 Anderson et al.. 1999 Peters et aI.. 2002 |
|
DFNA29 |
||
|
DFNA3() |
I 5q5-26 |
sconosciuto Mangino et al 200! |
|
DFNA3I |
6p2i.3 |
Sconosciuto |
Snoeckx et ai. 2004 |
|
DFNA32 |
i1p15 |
sconosciuto |
Li et ai.. 2000 |
|
DFNA33 |
13g34-gter |
sconosciuto |
Bonsch et ai.. 2009 |
|
DFA34 |
1g44 |
Kurinìa et ai., 2000 |
|
|
DFNA35 |
|||
|
DFNA36 |
9g13-g2i |
TMCI |
Kurimaetai.,2002 |
|
DFNA37 |
1p21 |
Taiebizadeh Ct ai.. 2000 |
|
|
DFA38 |
vedi DFNA6 |
Xiaoetai.. 2001 |
|
|
DFNA39 |
4g21.3 |
DSPP |
|
|
DFA40 |
i6pi2 |
||
|
DFNA4I |
12g24-gter |
sconosciuto |
Bianton_et_ai.,_2002 |
|
DFNA42 |
5g3 1.1 -g32 |
sconosciuto |
Xia et ai., 2002 |
|
DFNA43 |
2pi2 |
Fiex et ai,, 2003 |
|
|
DFA44 |
3q2829 |
CCDC5O |
Modarnio-Hoybjor et ai., 2003; Modamio-Hoybjor et ai. 2007 |
|
DFNA45 |
|||
|
DFNA46 |
|||
|
DFNA47 |
9p2i-22 |
sconosciuto |
D’Adamo et ai., 2003 |
|
DFNA$8 |
i2gi3-gi4 |
MYOiA |
D’Adamo et ai.. 2003. Donaudy et ai.. 2003 |
|
DFNA49 |
ig2i-g23 |
sconosciuto |
Moreno-Peiayo et ai.. 2003 |
|
DFNA50 |
7g32.2 |
MIRN96 |
Modamio-Hoybjor et ai.. 2004: Mencia et ai.. 2009 |
|
DFA5I |
9g2i |
TJP2 |
Waishetai.. 2010 |
|
DFNA52 |
4g28 |
||
|
DFN53 |
I 4g Il .2-g 12 |
sconosciuto |
Yan CI ai.. 2005 |
|
DFA54 |
5g3 I |
sconosciuto |
Gurtier et ai., 2004 |
|
DF>A55 |
|||
|
DFA56 |
|||
|
DFNA57 |
I9pI3.2 |
sconosciuto |
Bonsch et aI., 2008 |
|
DFNA5 |
2pi2-p2I |
sconosciuto |
Lezirovitz et ai.. 2009 |
|
DFNA59 |
i ipI4.2gi2.3 |
sconosciuto |
Chatterjee cI ai.. 2009 |
|
DFNA6(I |
2g2i.3-g24.i |
Liu XZet ai. ARO meeting. Denver. February 2007. |
|
Tabella III – Lista dei loci e dei geni delle ipoacusie non sindromiche recessive (da Van Camp e Smith 2010)
|
Locus |
Posizione Gene |
Referenze |
|
DFNB1A |
13q1 1-12 GJB2(in passato CX26) |
Guilford et al, 1994 |
|
Keiscll et al., 1997 |
||
|
DFNBIB |
13g12 GJB6(in passato CX3O) |
Del Castillo et aI., 2002 |
|
DFNB2 |
11q13.5 MYO7A |
— |
|
DFNB3 |
17pl 1.2 MYOI5A |
Fricdman et al., 1995 Wang et al., 1998 |
|
DFNB4 |
7q31 SLC26A4 |
Baldwin et al. 1995 Li Ct al., 1998 |
|
DFNB5 |
l4q12 sconosciuto |
Fukushima et al., 1995 |
|
(Vedi Nota 1) |
||
|
DFNB6 |
3p14—p21 TMIE |
Fukushima et al, 1995 Naz et al, 2002 |
|
DFNB7/1 I |
9g13-g21 TMCI |
Jain ci al.. 1995, Scoti ci al., l996,Kurima ci al.. 2002 |
|
DFNB8/DFNBIO |
21q22 TMPRSS3 |
Veskeetal., 1996, |
|
DFNB9 |
2p22-p23 OTOF |
Chaib ci al. 1996 |
|
(Vedi Nota 2) |
Yasunaga et al.. 1999 |
|
|
DFNBIO |
vedi |
|
|
DFNBI I |
vedi DFNB7 |
|
|
DFNBI2 |
10q21-q22 CDH23 |
Chaib ci al. 1996 Borketal.. 2001 |
|
DFNB 13 |
7g34-36sconosciuto |
Mustapha et al.. 1998 |
|
DFNBI4 |
7g31 sconosciuto |
Mustapha ci al.. 1998 |
|
DFNBI5 |
3q2l-q25 sconosciuto l9p13 |
Chen ci al, 1997 |
|
DFNB16 |
15g21-q22 STRC |
Campbcll ci al. 1997. Verpv ct al., 2001 |
|
DFNB17 |
7g31 Sconosciuto |
Greinwald ci al.. 1998 |
|
DFNB18 |
1 lpl4-l5.l USHIC |
Jain ci al, 1998. Ouyang ci aI., 2002; Ahmed ci al.. 2002 |
|
DFNB19 |
iSpi 1 sconosciuto |
The Molecular Biology of Hearing and Dcafness meeting Bcthcsda. October 8-11. 1998 (Green et al., abstract 108) |
|
DFNB2O |
1 lg25-gicr sconosciuto |
Moynihan ci al., 1999 |
|
DFNB2I |
I lg TECTA |
Mustapha ci al.. 1999 |
|
DFNB22 |
l6pl2.2 OTOA |
Zwaencpoel ci al.. 2002 |
|
DFNB23 |
lOpI l.2-q21 PCDI-115 |
Ahmed ci al. 2003 |
|
DFNB24 |
I lg23 RDX |
Khan et al.. 2007 |
|
DFNB25 |
4pl3 GRXCRI |
Schraders ci al.. 2010 |
|
DFNB26 |
4q3l sconosciuto |
Riazuddin Ct al,. 2000 |
|
(Nota 3) |
||
|
DFNB27 |
2g23-g3 I sconosciuto |
Pulleyn ci al.. 2000 |
|
DFNB28 |
22q13 TRIOBP |
Walsh ci al . 2000 |
|
DFNB29 |
21g22 CLDNI4 |
Wilcox ci al.. 2001 |
|
DFNB3O |
lOpI 1.1 MYO3A |
Walsh ci aI.. 2002 |
|
DFNB3 I |
9q32-q34 Wl-IRN |
Mustapha ci al . 2002 |
|
Mburu et al., 2003 |
||
|
DFNB32 |
lpl3.3-22.l GPSM2 |
Masmoudi et al., 2003; Walsh et al., 2010 |
|
DFNB33 |
9g34.3 sconosciuto |
Medlej-Hashim et aI., 2002 |
|
DFNB34 |
||
|
DFNB35 |
14q24.1- ESRRB |
Ansaret al., 2003: Collin et al,, 2008 |
|
DFNB36 |
I p36.3 ESPN |
Naz et al.. 2004 |
|
DFNB37 |
6g13 MYO6 |
Ahmedetal., 2003 |
|
DFNB38 |
6g26-g27 Sconosciuto |
Ansaret aL, 2003 |
|
DFNB39 |
7g21.l HGF |
Schultz etal., 2009 |
|
DFNB4O |
22g Sconosciuto |
Delmaghani et aI.. 2003 |
|
DFNB41 |
||
|
DFNB42 |
3q13.31- sconosciuto |
Aslam et aL, 2005 |
|
DFNB43 |
||
|
DFNB44 |
7pl4, 1- Sconosciuto |
Ansar et al., 2004 |
|
DFNB45 |
1g43-Q44 sconosciuto |
Bhatti et al., 2008 |
|
DFNB46 |
l8pll.32- sconosciuto plI.31 |
Miretal., 2005 |
|
DFNB47 |
2p2S,l- sconosciuto |
Hassan et al., 2005 |
|
DFNB48 |
15q23-g25.l sconosciuto |
Ahmad et al.. 2005 |
|
DFNB49 |
5q12 3- MARVELD2 |
Rainzan et al ,2004: Riazuddin et al ,2006 |
|
DFNB5O |
12g23 sconosciuto |
|
|
DF\B5I |
I 1p13-p12 sconosciuto |
Shaikh et al., 2005 |
|
DFNB52 |
||
|
DFNB53 |
6p2l.3 COLI 1A2 |
Chen et al., 2005 |
|
DFNB54 |
||
|
DFNB55 |
4g12-g13.2 sconosciuto |
Irshad et al., 2005 |
|
DFNB56 |
||
|
DFNB57 |
10q23.1- sconosciuto |
|
|
DFNB5S |
2q14 I- sconosciuto |
R Smith, inedito |
|
g21.2 |
||
|
DFNB59 |
2q3l 1- PJVK |
Delmaghani et al .2006 |
|
g31.3 |
||
|
DFNB6O |
5g22-g3 I Sconosciuto |
R. Smith. inedito |
|
DFNB6I |
7g22.I SLC26A5 |
Liu et al.. 2003 |
|
DFNB62 |
12pl3 2- sconosciuto |
Ali et al,, 2006 |
|
p1123 |
||
|
DFNB63 |
I Iq13.2- LRTOMT! COMT2 |
Du et al.. 2008: Ahmed et al., 2008 |
|
g13.4 |
||
|
DFNB6.i |
||
|
DFNB65 |
20q13 2- sconosciuto |
Tariq et al . 2006 |
|
g13.32 |
||
|
DFNB66 67 |
6p2l.2-22.3 LHFPL5 |
TIiIi Ct aI. 2005. Shabbiret al.. 2006: Kalay et al., 2006 |
|
DFB6 |
Vedi |
|
|
DFNB66 |
||
|
DFNB6S |
l9pl3.2 sconosciuto |
Santos et al.. 2006 |
|
DFNB69 |
Nota 1. DFNB5 è stata segnalata originariamente come DFNB4.
Nota 2. DFNB9 è stato segnalato in origine come DFNB6.
Nota 3. DFNB26 è soppresso da modificatore dominante DFNM1.
Esistono molti tipi di test genetici disponibili che colpiscono diverse porzioni del genoma. I disordini genetici possono essere causati da anomalie nelle varie regioni; il metodo per identificare il difetto genetico dipende dal tipo di sindrome genetica o anormalità sospettata e, quindi, la loro nota difetto genomico. Da larga scala per piccoli cromosoma o DNA modifiche, disturbi genetici possono derivare da una variazione del numero di cromosomi, delezione o l’aggiunta di una porzione di una regione cromosomica o una mutazione del DNA all’interno di un singolo gene.
Citogenetica (cariotipo o cromosoma) test viene utilizzato per rilevare anomalie cromosomiche. Le variazioni del numero di cromosomi in ogni cellula sono spesso incompatibili con la vita, perché ogni cromosoma contiene migliaia di geni. Perdita o guadagno di un numero così elevato di geni è molto dannoso per lo sviluppo. La forma più comune di sopravvivere cromosoma numero aberrante è la sindrome di Down, che colpisce 1 su 700 nascite (Ramia, Musharrafieh, Khaddage, e Sabri, 2013). La sindrome di Down, detta anche Trisomia 21 , i risultati di una copia in più del cromosoma 21. Gli individui con sindrome di Down, quindi, hanno un numero di cromosomi di 47 invece del solito 46. sindrome di Down è una delle principali cause di disabilità intellettiva e deficit dell’udito sono riportati nel 34% al 78% dei casi pediatrici (Raut et al, 2011;. Shott, Joseph, e Heithaus, 2001). La perdita dell’udito può essere conduttiva, neurosensoriale, o misto (Austeng et al., 2013), e l’otite media è molto comune (Ramia et al., 2013). La maggior parte dei casi di sindrome di Down sono sporadici, ma un piccolo numero di casi sono ereditate (Verma, Lall, e Dua Puri, 2012). L’età avanzata materna è un importante fattore di rischio (Smith & Visootsak, 2013). La diagnosi viene effettuata mediante analisi cromosomica di sangue o, nel periodo prenatale, per l’analisi cromosomica delle amniotico prelievo dei villi coriali o liquido. I campioni sono esaminati al microscopio per analizzare il numero totale di cromosomi presenti e per determinare quali cromosomi sono duplicati o mancanti. Le cellule vengono esposte e cromosomi sono disposti in un cariotipo (vedi Figura 5). Test citogenetica è estremamente utile nella diagnosi di trisomie e altre anomalie cromosomiche. Tuttavia, riarrangiamenti genetici più piccole potrebbero non essere visibili a causa della sua limitata risoluzione.
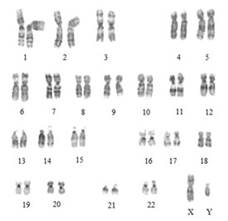
Figura 5. cariotipo maschile normale
I cromosomi con delezioni o duplicazioni troppo piccoli per essere visualizzati con i test citogenetici di routine può essere ulteriormente analizzate da FISH (a fluorescenza in situ ibridizzazione). test FISH comporta l’applicazione di un breve segmento di DNA (sonda) che codifica per il disturbo sospetto marcato con un tag fluorescenti . La sonda a fluorescenza si collega alla sua sequenza complementare sul campione del paziente. Le cellule sono poi analizzati per la presenza o l’assenza del segnale di sonda fluorescente al microscopio. Il vantaggio di FISH è che è molto specifico per la diagnosi di microdeletions e microduplicazioni. Tuttavia, un disturbo particolare deve essere sospettata in modo che venga utilizzato la sonda FISH corretta. Un elenco di sindromi microdelezione si possono trovare sul sito web Springer Protocols (Schwartz & Graf, 2002). Un esempio di un saggio FISH è mostrata in Figura 6.
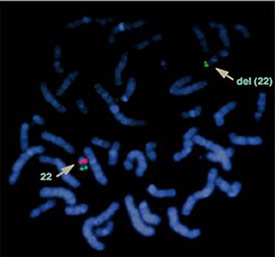
Figura 6. La diagnosi di sindrome di DiGeorge utilizzando la fluorescenza in situ ibridazione (FISH).
Segnale verde: normale controllo. Segnale Rosso: sindrome di DiGeorge regione del gene. Del (22): la cancellazione positiva del gene sul cromosoma 22 DiGeorge.
Negli ultimi anni, la tecnologia microarray è stata sviluppata per test genetici. Microarray ibridazione genomica comparativa (CGH) confronta il DNA dal paziente con DNA da un controllo normale. I campioni di DNA del paziente e di controllo sono etichettati con sonde fluorescenti di colore diverso e lasciati a competere per il legame alle migliaia di segmenti di DNA su un vetro “chip”. Questo viene poi analizzato da un laser e le differenze tra il DNA paziente e normale DNA di controllo vengono analizzati da un software specializzato. Gli utili e le perdite di DNA possono essere visualizzati da questa tecnologia, che offre un ampio panorama dell’intero genoma; è, quindi, non è necessario conoscere l’anomalia genetica esatto in cui si richiede questo test. Microarrays possono essere personalizzati per la ricerca di un particolare gruppo di disturbi. Ad esempio, un chip con segmenti di molti geni differenti associati con perdita di udito può essere creato per testare più geni sordità contemporaneamente. Una limitazione di microarray è risultato insolito che possono essere difficili da interpretare e le differenze tra una variante genetica normale e una vera patologia genetica (Evangelidou et al., 2013).
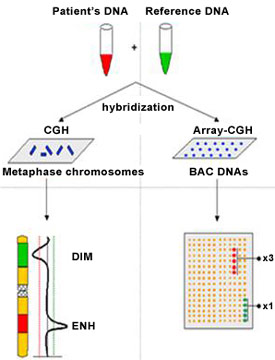
Figura 7. microarray Comparative Genomic Hybridization (CGH)
Alcuni tipi di perdita dell’udito sono causate da mutazioni o cambiamenti nel DNA che sono troppo piccole per essere rilevate dai metodi precedentemente menzionati. La mutazione analisi può essere effettuata da PCR (polymerase chain reaction ). PCR è una tecnica di amplificazione di un piccolo segmento di DNA per produrre milioni di copie. Questa tecnica genera un ampio campione di DNA che può essere analizzato visualizzando il campione attraverso elettroforesi su gel. Il cambiamento genetico più piccolo comporta la sostituzione di una coppia di basi di DNA con un altro, definita una mutazione puntiforme .Trovare una mutazione puntiforme può anche richiedere sequenziamento del DNA del gene in questione dopo PCR. La tecnica di sequenziamento del DNA prevede l’analisi di ogni base DNA della regione del gene, consentendo il rilevamento di sostituzioni, delezioni o inserzioni, comportano una singola base. Gene sequenziamento è il metodo preferito di prove per Connexin 26 mutazioni (Figura 8; Angeli et al, 2012.).
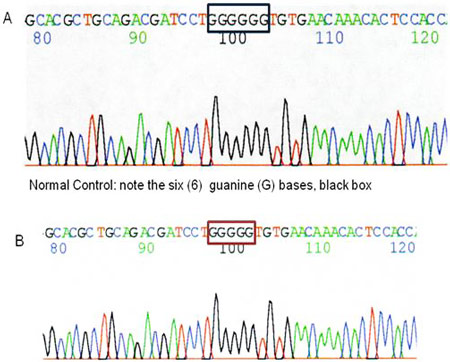
Figura 8. La diagnosi di connessina 26 mutazioni dal sequenziamento del DNA.
Risultati positivi per la sordità causata da connessina 26. Poiché questo tipo di sordità segue un modello di eredità autosomica recessiva, un paziente affetto dimostra una delezione di una guanina (G) di base in entrambi Connexin 26 (GJB2) geni. Notare le cinque basi (5) G al posto della normale sei (6), scatola rossa.
Oltre alla diagnosi del paziente, tecniche genetiche sono cruciali per l’udienza di ricerca perdite. I geni umani possono essere inseriti in altri organismi di studiare come funzionano. I topi sono gli organismi di scelta nello studio dei geni sordità per le seguenti ragioni: (1) il genoma e orecchio interno del topo sono molto simile a quello umano, (2) topi hanno un breve tempo di gestazione, e (3) sono relativamente facili da accoppiarsi selettivamente. (2012 Angeli et al., Ni et al, 2013). Numerosi modelli di topo che rappresentano modelli in perdita compresa udito umano per la sindrome di Usher e Waardenburg syndrome- sono stati sviluppati.
Sommario
Molti diversi tipi di cambiamenti genetici possono essere associati con la perdita dell’udito. Il tipo di test diagnostico selezionato dipenderà dal tipo di cambiamenti genetici sospetti. Gli indizi possono essere dedotte dal modello storia di famiglia e di successione, ma i medici devono ricordare che una storia familiare negativa non implica che l’eziologia non è genetica. I test genetici può permettere per la diagnosi precoce di altri potenziali problemi di salute, valutare i rischi di ricorrenza, e avvisare gli altri membri della famiglia che possono essere interessati. I test genetici non dovrebbe essere intrapreso fino a cause ambientali, come le infezioni e traumi, sono escluse. Audiologi dovrebbero individuare i genetisti nella loro area e mantenere uno stretto rapporto con loro per la cura ottimale dei pazienti con forme genetiche di perdita dell’udito. Informazioni su sindromi genetiche, le prove, e genetisti nella vostra zona si possono trovare i National Institutes of Health Test genetici Registry e GeneTests siti web.
Ahmed, Z. M., Riazuddin, S., Riazuddin, S., & Wilcox, E. R. (2003). The molecular genetics of Usher syndrome. Clinical Genetics, 63, 431–444.
Angeli, S., Lin, X., & Liu, X. Z. (2012). Genetics of hearing and deafness. Anatomical Record, 295,1812–1829.
Austeng, M. E., Akre, H., Overland, B., Abdelnoor, M., Falkenberg, E. S., & Kvaerner, K. J. (2013). Hearing level in children with Down syndrome at the age of eight. Research in Developmental Disabilities, 34, 2251–2256.
Chen, G., He, F., Fu, S., & Dong, J. (2011). GJB2 and mitochondrial DNA 1555A>G mutations in students with hearing loss in the Hubei province of China. International Journal of Pediatric Otorhinolaryngology, 75, 1156–1159.
de Sousa Andrade, S. M., Monteiro, A. R., Martins, J. H., Alves, M. C., Santos Silva, L. F.,
Quadros, J. M., & Ribeiro, C. A. (2012). Cochlear implant rehabilitation outcomes in Waardenburg syndrome children. International Journal of Pediatric Otorhinolaryngology, 76, 1375–1378.
Evangelidou P., Alexandrou, A., Moutafi, M., Ioannides, M., Antoniou, P., Koumbaris, … Patsalis, P. C. (2013). Implementation of high resolution whole genome array CGH in the prenatal clinical setting: Advantages, challenges, and review of the literature. BioMed Research International, 2013, 346762.
Guha S., Rosenfeld, J. A., Malhotra, A. K., Lee, A. T., Gregersen, P. K., Kane, J. M., … Lencz, T. (2012). Implications for health and disease in the genetic signature of the Ashkenazi Jewish population. Genome Biology, 13, R2.
Hilgert, N., Smith, R. J. H., & Van Camp, G. (2009). Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutation Research, 681, 189–196.
Keats, B. J. (2002). Genes and syndromic hearing loss. Journal of Communication Disorders, 35, 355–366.
Kenneson, A., Van Naarden Braun, K., & Boyle, C. (2002). GJB2 (Connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genetics in Medicine, 4, 258–274.
Kruegel, J., Rubel, D., & Gross, O. (2013). Alport syndrome- insights from basic and clinical research.Nature Reviews Nephrology, 9, 170–178.
Liu, F., Li, P., Liu, Y., Li, W., Wong, F., Du, R., & … Liu, M. (2013). Novel compound heterozygous mutations in MYO7A in a Chinese family with Usher syndrome type 1. Molecular Vision, 19, 695–701.
National Center for Hearing Assessment and Management. (2010). Universal newborn hearing screening: Issues and evidence. Retrieved April 28, 2013.
Ni, C., Zhang, D., Beyer, L. A., Halsey, K. E., Fukui, H., Raphael, Y., & … Hornyak, T. J. (2013). Hearing dysfunction in herterozygous Mitf(Mi-wh)/+ mice, a model for Waardenburg syndrome type 2 and Tietz syndrome. Pigment Cell Melanoma Research, 26, 78–87.
Ouyang, X. M., Hejtmancik, J. F., Jacobson, S. G., Xia, X. J., Li, A., Du, L. L., & … Liu, X. Z. (2003). USH1C: a rare cause of USH1 in a non-Acadian population and a founder effect of the Acadian allele.Clinical Genetics, 63, 150–153.
Ramia, M., Musharrafieh, U., Khaddage, W., & Sabri, A. (2013). Revisiting Down syndrome from the ENT perspective: Review of literature and recommendations [published ahead of print, May 21]. European Archives of Oto-Rhino-Laryngology.
Raut, P., Sriram, B., Yeoh, A., Hee, K. Y., Lim, S. B., & Daniel, M. L. (2011). High prevalence of hearing loss in Down syndrome at first year of life. Annals of the Academy of Medicine, Singapore, 40, 493–498.
Read, A. P., & Newton, V. E. (1997). Waardenburg syndrome. Journal of Medical Genetics, 34, 656–665.
Rehm, H. L. (2005). A genetic approach to the child with sensorineural hearing loss. Seminars in Perinatology, 29, 173–181.
Rheault, M. N. (2012). Women and Alport syndrome. Pediatric Nephrology, 27, 41–46.
Schwartz, S., & Graf, M. D. (2002). Microdeletion syndromes: Characteristics and diagnosis. In Y. S. Fan (Ed.), Molecular cytogenetics: Protocols and applications (pp. 275–290). Retrieved October 3, 2013.
Shott, S. R., Joseph, A., & Heithaus, D. (2001). Hearing loss in children with Down syndrome.International Journal of Pediatric Otorhinolaryngology, 61, 199–205.
Smith, M., & Visootsak, J. (2013). Noninvasive screening tools for Down syndrome: a review.International Journal of Women’s Health, 5, 125–131.
Smith, R. J., Bale, Jr., J. F., & White, K. R. (2005). Sensorineural hearing loss in children. The Lancet, 365, 879–890.
Toriello, H. V., Reardon, W., & Gorlin, R. J. (2004). Hereditary hearing loss and its
syndromes. New York, NY: Oxford University Press.
Van Camp, G., & Smith, R. J. H. (2000). Maternally inherited hearing impairment. Clinical Genetics, 57,409–414.
Van Camp, G., & Smith, R. (2013). Hereditary hearing loss homepage. Retrieved July 31, 2013.
Verma, I. C., Lall, M., & Dua Puri, R. (2012). Down syndrome in India—diagnosis, screening, and prenatal diagnosis. Clinics in Laboratory Medicine, 32, 231–248.
Yan, D., & Liu, X. Z. (2010). Genetics and pathological mechanisms of Usher syndrome. Journal of Human Genetics, 55, 327–335.