TRASMISSIONE IPOACUSIA AUTOSOMICA DOMINANTE (AD) (22%)
In un modello di ereditarietà autosomica dominante , un bambino eredita una copia normale di un gene da un genitore e un gene anomalo dall’altro genitore. Nelle famiglie affette da sordità autosomica dominante, uno dei genitori è sordo e porta su un singolo allele del gene la mutazione patogena, che trasmetterà a metà dei suoi figli, che quindi saranno sordi. Il gene anomalo domina il gene normale, così una copia di un gene anormale è sufficiente a causare una malattia autosomica dominante (Fig. 1).La mutazione dell’allele interessato può. per esempio. produrre una proteina anormale, che impedisce la funzione della proteina normale prodotta dall’allele sano. La Sindrome di Waardenburg è la causa più comune di perdita di udito sindromica autosomica dominante, che colpisce 1 su 42.000 persone (Read & Newton, 1997). Quando un genitore ha la sindrome di Waardenburg, ogni bambino di quel genitore ha una probabilità del 50% di essere affetti da sindrome di Waardenburg, assumendo l’altro genitore non ha la sindrome. Il restante 50% della prole in questo accoppiamento sarà inalterato. Caratteristiche tipiche della sindrome di Waardenburg includono perdita dell’udito neurosensoriale, un ciuffo bianco, occhi celesti o di colore diverso, e gli occhi distanziati. Una grande quantità di espressività variabile esiste in Waardenburg sindrome, in modo che i pazienti possono avere una qualsiasi combinazione di caratteristiche, tra cui un udito normale (de Sousa Andrade et al., 2012). Waardenburg sindrome rappresenta circa il 2% dei casi di sordità profonda congenita (de Sousa Andrade et al., 2012).Va notato che, in rari casi, alcuni disturbi autosomica dominante verificarsi a causa di una nuova mutazione nel bambino e nessuno dei genitori ha il disturbo. L’espressività è però variabile in questa modalità di trasmissione: la gravità della ipoacusia può essere variabile tra i vari soggetti colpiti.
Quando l’ipoacusia è sindromica i segni associati alla sindrome possono essere assenti o lievi in alcuni sordi della famiglia e alcuni membri portatori dell’ alterazione genetica possono essere udenti.
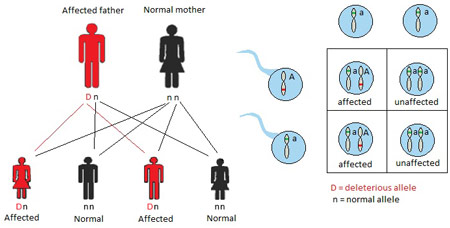

Fig.1 ereditarietà autosomica dominante
Le sordità sindromiche rappresentano solo una piccola percentuale di sordità del bambino (10 – 15% circa) e una porzione poco conosciuta, probabilmente inferiore, di sordità dell’adulto. Sono state descritte diverse centinaia di sindromi con sordità (vedi [Gorlin et al.,1995 8]), e a tutt’oggi è stato identificato più di un centinaio di geni. È comunque importante conoscere e ricercare le principali sindromi, poiché la gestione e la valutazione eziologica saranno differenti da una sordità non sindromica. In ragione del grande numero di sindromi rare con sordità qualsiasi patologia malformativa nel bambino deve far effettuare un esame uditivo sistematico. Per le sordità sindromiche inoltre che comprendono una lesione malformativa craniofaciale, la sordità è molto spesso aggravata da un’otite cronica e s’impone un monitoraggio otologico regolare.
Abbiamo elencato nella Tabella 4 le sette sordità sindromiche che ci sembrano più importanti e che devono essere conosciute dai diversi specialisti che assumono in carico le sordità, a motivo della loro frequenza e/o della loro gravità potenziale. La Tabella 5 elenca in modo più completo le sindromi no eccezionali i cui geni sono stati identificati (per una rassegna esauriente dei geni clonati della sordità sindromica, vedi [Marlin S. 2004 9]). Descriveremo qui le sette sindromi della Tabella 1.
Tre sindromi autosomiche dominanti
Devono essere conosciute dagli otorinolaringoiatri (ORL) poiché sono frequenti e probabilmente sottodiagnosticate: la sindrome di Waardernburg, la sindrome branchio-oto-renale (BOR) e la sindrome di Stickler.
Associa una sordità ad anomalie della pigmentazione dovute a un’assenza di melanociti in diversi organi. Ciò può riguardare i capelli (ciocche bianche) e le sopracciglia, gli occhi (occhi molto azzurri, depigmentati, depigmentazione sul fondo dell’occhio), la cute (macchie cutanee). In alcune forme (Waardernburg di tipo 1 e 3), è presente un’anomala distanza tra gli occhi, con i canti interni spostati verso l’esterno con riduzione di lunghezza della fessura palpebrale (distopia cantale). La sordità è molto variabile, mono- o bilaterale, da leggera a profonda (profonda nel 35% dei casi), [Hildesheimer et al.,1989 24] con o senza malformazione dell’orecchio interno. La trappola per la diagnosi è che le particolarità fisiche che possono essere presenti nella famiglia dei sordi ed essere molto suggestive non sono spontaneamente descritte all’anamnesi, non essendo considerate patologie. Le ciocche bianche sono inoltre il più delle volte tinte. La domanda circa la presenza di occhi depigmentati o di ciocche bianche nella famiglia deve quindi essere posta sistematicamente. Sono stati descritti quattro tipi clinici di sindrome di Waardernburg in funzione dei segni associati:
• il tipo I è associato a una distopia cantale;
• il tipo II, il più frequente, è privo di distopia cantale;
• il tipo III (o Klein-Waardenburg) è un tipo I associato a malformazioni delle estremità;
• il tipo IV è un tipo II con malattia di Hirschsprung. Attualmente, sono stati identificati sei geni, onnicomprensivi nella Tabella 4. Essi non rendono conto di tutte le sindromi di Waardernburg (per esempio, PAX3 è chiamato in causa in tre quarti dei tipi I e MITF nel 15% dei tipi II soltanto). [Tassabehjiet al.,1995 25]
Sindrome branchio-oto-renale
La sindrome branchio-oto-renale (BOR) associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000. [Fraser et al.,1980 26] Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un’interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un’ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell’orecchio esterno (malformazioni del padiglione, aplasia dell’orecchio, encondromi, stenosi dei condotti uditivi), dell’orecchio medio (esiste una componente di trasmissione all’audiogramma) e dell’orecchio interno (varie malformazioni cocleovestibolari). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi). In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell’orecchio esterno, è consigliabile eseguire un’ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1. [Abdelhak et al.,1997;, Ruf et al.,2004 28] Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale. [Vincent et al., 1997 29]
Sindrome di Stickler
Questa sindrome è dovuta a una alterazione delle catene ![]() di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
di alcuni collageni. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le anomalie articolari possono portare a dolori di tipo artrosico.
La sordità di percezione, trasmissione o mista è incostante, e spesso mascherata o aggravata dai problemi di otite cronica che sono associati all’incompetenza faringea. Essa è spesso evolutiva. Sono descritti tre tipi di sindrome di Stickler: nei tipi 1 e 3, agli altri elementi della sindrome è associata una miopia molto forte con rischio di degenerazione vitro-retinica. L’esame oftalmologico di ogni bambino sordo e di ogni lattante con incompetenza faringolaringea deve permettere d’individuare questa sindrome e correggere precocemente la miopia.
Gorlin R.J., Toriello H.V., Cohen M.M. Hereditary hearing loss and its syndromes New York: Oxford University Press (1995).
Marlin S. Surdités génétiques Génétique médicale Paris: Masson (2004). 281-299
Hildesheimer M., Maayan Z., Muchnik C., Rubinstein M., Goodman R.M. Auditory and vestibular findings in Waardenburg’s type II syndrome J. Laryngol. Otol. 1989 ; 103 : 1130-1133
Tassabehji M., Newton V.E., Liu X.Z., Brady A., Donnai D., Krajewska-Walasek M., e al. The mutational spectrum in Waardenburg syndrome Hum. Mol. Genet. 1995 ; 4 : 2131-2137
Fraser F.C., Sproule J.R., Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss Am. J. Med. Genet. 1980 ; 7 : 341-349 [cross-ref]
Abdelhak S., Kalatzis V., Heilig R., Compain S., Samson D., Vincent C., e al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family Nat. Genet. 1997 ; 15 : 157-164 [cross-ref]
Ruf R.G., Xu P.X., Silvius D., Otto E.A., Beekmann F., Muerb U.T., e al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes Proc. Natl. Acad. Sci. USA 2004 ; 101 : 8090-8095 [cross-ref]
Vincent C., Kalatzis V., Abdelhak S., Chaib H., Compain S., Helias J., e al. BOR and BO syndromes are allelic defects of EYA1 Eur. J. Hum. Genet. 1997 ; 5 : 242-246
APPROFONDIMENTO
Waardenburg sindrome (anche sindrome di Shah Waardenburg, sindrome di Waardenburg-Klein, sindrome di Mende II, Van der sindrome Hoeve-Halbertsma-Waardenburg, sindrome di ptosi-epicanto, Van der sindrome Hoeve-Halbertsma-Gualdi, tipo Waardenburg Pierpont, [5] Van der Hoeve sindrome Waardenburg-Klein, sindrome di Waardenburg II e sindrome di Vogt) è una rara malattia genetica più spesso caratterizzato da diversi gradi di sordità , difetti minori nelle strutture derivanti dalla cresta neurale , e anomalie della pigmentazione. Disagi Nella miogenesi , in particolare mutazioni in Pax3, possono causare la sindrome di Waardenburg I e III.E ‘stata descritta per la prima nel 1951.

 Che cosa è la sindrome di Waardenburg?,
Che cosa è la sindrome di Waardenburg?,
Le cause della sindrome di Waardenburg,
Fisiopatologia,
Epidemiologia,
Diagnosi e valutazione audiologica,
Che cosa è la sindrome di Waardenburg?
Waardenburg sindrome è una malattia rara caratterizzata da sordità in associazione con anomalie della pigmentazione e difetti dei tessuti cresta di derivazione neurale.
Sindrome è una parola che descrive un gruppo di caratteristiche fisiche che si verificano insieme. Le persone che hanno WS di solito hanno un aspetto unico. Questa unicità è causato da cambiamenti nel loro corredo genetico.
Sindrome di Waardenburg è una condizione genetica che causa una persona di essere nato con le caratteristiche facciali inusuali e perdita dell’udito. Questi problemi fisici possono comparire nel corso della vita della persona. Non tutti con WS avrà tutte le caratteristiche della sindrome, e non tutti avranno una perdita uditiva, ma vi è una maggiore probabilità che lo faranno.
Hammerschlag, nel 1907, e Urbantschitsch, nel 1910, entrambi citano eterocromia dell’iride, albinismo parziale con complicanze di sordomutismo Nel 1916, van der Hoeve descrive una distopia angoli palpebrali mediale lateroversa in una coppia di gemelle monozigoti con Sordomutismo La Sindrome di Waardenburg è stato identificato nel 1951 da Petrus Johannes Waardenburg(1886-1979), un oculista olandese che ha notato che molti dei suoi pazienti avevano caratteristiche facciali uniche, occhi di colore diverso avevano pure, spesso, la perdita dell’udito. In effetti, una caratteristica primaria di Waardenburg è perdita di udito – insieme con gli occhi blu/ marroni o due brillanti occhi azzurri. Un’altra caratteristica facilmente riconoscibile è capelli bianchi sulla testa. Da allora, sono stati descritti quattro tipi specifici di WS. La condizione ha descritto è ora classificato come WS1.
ha definito la sindrome con le seguenti 6 principali caratteristiche
· Spostamento laterale della angoli palpebrali mediali combinata con distopia del punto lacrimale e blefarofimosi
· Aspetto di Eyes Wide-set a causa di una prominente, larga radice del naso(distopia dei canti ),associata in particolare con il tipo I),nota anche come telecanto ;
· Ipertricosi della parte mediale delle sopracciglia che toccano nel mezzo
· Un ciuffo di capelli bianchi ( poliosi ), o ingrigimento precoce dei capelli;
· Occhi molto chiari o azzurri brillanti, occhi di due colori diversi (eterocromia completa ), o gli occhi con un’iride con due colori diversi (eterocromia settoriale);
· Sordità da moderata a profonda (frequenza più alta associata al tipo II);
L’oculista svizzero David Klein, nel 1947, ha anche fatto contributi per la comprensione della sindrome,Klein ha riportato il caso di una ragazza di 10 anni con deafmutism, albinismo parziale della pelle e dei capelli, iridis ipocromia, blefarofimosi con ipertelorismo e l’assenza dell’angolo nasofrontal, ipertricosi delle sopracciglia, e molteplici anomalie associate (mio -osteo-Articulare displasia).
WS2 è stato identificato nel 1971, per descrivere i casi di ” distopia dei canti “non era presente. [4] WS2 è ora diviso in sottotipi, basato sul gene responsabile.
Sono stati identificati altri tipi, ma sono meno comuni.
Mutazioni nel EDN3, EDNRB, MITF, PAX3, SNAI2, e SOX10 sono i geni che sono affetti dalla sindrome di Waardenburg. Alcuni di questi geni sono coinvolti nella realizzazione dei melanociti, che rende il pigmento melanina. La melanina è un pigmento importante nello sviluppo di capelli, il colore degli occhi, della pelle, e le funzioni dell’orecchio interno. Così la mutazione di questi geni può portare a pigmentazione anomala e la perdita dell’udito. [5] PAX3 e MTIF mutazione del gene avviene in tipo I e II (WS1 e WS2). Tipo III (WS3) mostra mutazioni del gene PAX3 anche. Mutazioni del gene SOX10, EDN3, o EDNRB si verificano in tipo IV.Tipo IV (WS4) può anche interessare porzioni di sviluppo delle cellule nervose che potenzialmente possono portare a problemi intestinali.
Le cause della sindrome di Waardenburg
Questa condizione è solitamente ereditata come autosomica dominante modello, il che significa una copia del gene alterato è sufficiente a causare il disturbo. Nella maggior parte dei casi, una persona affetta ha un genitore con la condizione. Una piccola percentuale di casi derivano da nuove mutazioni nel gene; questi casi si verificano in persone senza storia di malattia nella loro famiglia.
Alcuni casi di tipo II e sindrome tipo IV Waardenburg sembrano avere un autosomica recessiva modello di ereditarietà, il che significa due copie del gene devono essere modificati per una persona di essere colpiti dalla malattia. Molto spesso, i genitori di un bambino con una malattia autosomica recessiva non sono interessati, ma sono portatori di una sola copia del gene alterato.
· 
Waardenburg sindrome è solitamente ereditata come modello autosomico dominante .
· 
Tipi II e sindrome IV Waardenburg possono talvolta avere un modello di ereditarietà autosomico recessivo .
I geni sono responsabili per dirigere la crescita e lo sviluppo degli esseri umani. Quando i geni sono mutati, o diverso dal normale, possono causare cambiamenti nella normale crescita e sviluppo. In WS, i geni mutati sono responsabili di distribuzione del pigmento nel corpo di sviluppo embrionale e fetale. Questi geni possono causare pigmento da distribuire in modo anomalo. Quando questo accade, alcune zone del corpo possono avere troppo o troppo poco pigmento, o nessun pigmento affatto. Alterazioni della pigmentazione possono produrre occhio inusuale, la pelle, o il colore dei capelli così come la sordità. Gli scienziati non hanno ancora capito esattamente che cosa causa le mutazioni genetiche che producono WS, anche se sono stati identificati i geni stessi.
Fisiopatologia
La sindrome Waardenburg è una malattia rara con una modalità autosomica dominante. Diverse ipotesi sono state avanzate per spiegare tutte le caratteristiche cliniche della sindrome.
- La teoria della cresta neurale carente, suggerisce una anomalia dello sviluppo della cresta neurale come causa di malattia: L’associazione della sindrome di Waardenburg e megacolon congenito aganglionico supporta questa ipotesi.
- La sindrome di Waardenburg come una parte della sindrome del primo arco
- Un rapporto della sindrome di Waardenburg con lo status dysraphicus
- La teoria della necrosi intrauterina
Nessuna di queste possibilità spiega tutte le caratteristiche della sindrome di Waardenburg. Cause ereditarie rappresentano circa il 50% degli individui osservati per l’infanzia (prelinguale) perdita di udito, di cui il 70% sono dovuti a mutazioni in numerosi geni singoli che compromettono la funzione uditiva solo (non sindromica). Il resto sono associate ad altre anomalie dello sviluppo chiamati sordità sindromica.
I Geni responsabili di forme sindromiche di perdita dell’udito nella sindrome di Waardenburg includono PAX3 sulla banda 2q37, osservata nei tipi I e III, e MITFmappato su 3p12-p 14,1 per il tipo II(Tachibana M.,1997; Zhang H et al., 2012; Wildhardt G et al 2013 . La sindrome di Waardenburg è autosomica dominante per la maggior parte delle persone con tipo I, II, e III. La sindrome di Waardenburg di tipo IV è autosomica recessiva con penetranza variabile ed è causa di mutazioni del gene SOX10 o del recettore dell’endotelina-B ( EDNRB ), che sembrano correlare con l’intestinale e / o sintomi neurologici manifesta nei pazienti(Morell R, et al.1998;DeStefano AL, et al.,1998;Bondurand N, et al.,2000; Dundar M, et al.,2001;Chan KK, et al.,2003; Sznajer et al.,2008; Oshimo T, et al.,2012;Jung HJ, et al.,2013;hanno descritto un sito di splice mutazione SOX10 (c.698-2A> C), che ha provocato una sindrome di Waardenburg grave di tipo 4 senza malattia di Hirschsprung . Il bambino presentava vivaci occhi blu, ritardo mentale, sinofria, sordità, canali completi bilaterali semicircolari, e neuropatia periferica.
Epidemiologia
Quanto rara è la sindrome di Waardenburg?
La frequenza della sindrome Waardenburg è stimato a 1 caso ogni 212.000 persone nella popolazione generale dei Paesi Bassi, ma a causa di una bassa penetranza di circa il 20%, la frequenza dell’intero sindrome (con o senza sordità) è probabilmente di circa 1 caso per ogni 42.000 persone. In Kenya, Secondo il National Institutes of Health, un individuo su 10.000 a 20.000 avrà la sindrome di Waardenburg. Inoltre, dal momento che i primi due tipi (tipo I e II) sono più comuni e sono associati con la perdita dell’udito, si può presumere che la maggior parte degli individui con Waardenburg avrà la perdita dell’udito.
La sindrome è stata osservata in 0,9-2,8% delle persone con sordomutismo.
Mortalità / morbilità
I bambini con sindrome di Waardenburg hanno una aspettativa di vita normale. La morbilità è legato alla sordità e difetti dei tessuti cresta di derivazione neurale, tra cui ritardo mentale, convulsioni, disturbi psichiatrici, anomalie scheletriche, e disturbi oculari (cataratta comprese).
Razze
Waardenburg sindrome colpisce persone di tutte le razze di tutto il mondo.
Sesso
La malattia colpisce ugualmente entrambi i sessi. Non sono stati trovati differenze sessuali tra persone con sordomutismo congenito.
Età
Come una malattia ereditaria, la sindrome di Waardenburg può essere riconosciuto immediatamente o subito dopo la nascita. Alcune caratteristiche dermatologiche (ad esempio, poliosi) cambiano con l’età.
Chi è a rischio?
Si stima che circa 1 su 4000 individui nascono con WS. WS si verifica in tutte le razze, e si verifica in modo uguale in maschi e femmine. Nella maggior parte dei tipi di WS, c’è una probabilità del 50% che un bambino nato da una madre con un gene normale e un genitore con il gene WS avrà WS. Chiunque può nascere con WS come risultato di una nuova mutazione genetica.
Il trattamento dipende dal fenotipo dell’individuo con Sindrome di Waardenburg. È possibile ottenere un trattamento per uno dei seguenti segni fenotipiche:
1) Sordità da moderata a profonda (frequenza più alta associata al tipo II);
2) La malattia di Hirschsprung (HD)
3) problemi estetici
4) palatoschisi
5) Mano e contratture delle dita
Il trattamento per queste caratteristiche comporta la consulenza genetica e / o rinvio di appropriarsi medici professionisti come un audiologo, logopedista, fisioterapista e chirurgo plastico.
Diagnosi e valutazione audiologica
Americans with Disabilities Act (ADA)
La perdita dell’udito può essere trattata, l’udito deve essere controllato da un audiologo. Lui o lei determinerà il grado e il tipo di perdita uditiva. Ecco un elenco di alcuni dei test che un audiologo può somministrare. Non tutti hanno bisogno di tutti questi test. Il medico o l’audiologo può raccomandare quelli necessari, in base alla storia familiare ei sintomi della perdita dell’udito.
Valutazione audiologica
1. Otoscopia
 L’Otoscopia è un esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di infezione.
L’Otoscopia è un esame fisico del vostro orecchio esterno, condotto uditivo, e timpano. Un audiologo userà un otoscopio per cercare eventuali perforazioni del timpano, arrossamento del timpano, cerume o segni di infezione.
2. Timpanometria
 La timpanometria è una misura della rigidità del timpano e ci dice come gli ossicini dell’orecchio medio funzionano. Questo test consente di rilevare liquido nell’orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si trovano nell’orecchio medio, un perforazione nel timpano, e una malattia dell’orecchio medio chiamato otosclerosi. L’audiologo metterà una sonda morbida nel canale uditivo e una macchina rilascia una piccola quantità di pressione. Il movimento strumento misura le risposte del timpano alle variazioni di pressione e registra il risultato su un grafico.
La timpanometria è una misura della rigidità del timpano e ci dice come gli ossicini dell’orecchio medio funzionano. Questo test consente di rilevare liquido nell’orecchio medio, rottura o eventuali lussazioni degli ossicini ,che si trovano nell’orecchio medio, un perforazione nel timpano, e una malattia dell’orecchio medio chiamato otosclerosi. L’audiologo metterà una sonda morbida nel canale uditivo e una macchina rilascia una piccola quantità di pressione. Il movimento strumento misura le risposte del timpano alle variazioni di pressione e registra il risultato su un grafico.
3. Audiometria Tonale ai Toni Puri
 Il test ai toni puri è il tipo più comune di valutazione dell’udito. L’audiologo può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il suono. L’audiologo registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri comprende anche il test di conduzione per via ossea. Durante questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e determinare quale parte dell’orecchio è responsabile di qualsiasi perdita uditiva.
Il test ai toni puri è il tipo più comune di valutazione dell’udito. L’audiologo può utilizzare cuffie o inserire gli auricolari, che sono come tappi per le orecchie, per la valutazione della conduzione per via aerea. Durante il test, si sentono toni a diverse frequenza ed intensità, e l’esaminato deve alzare la mano quando sente il suono. L’audiologo registra le soglie per ogni frequenza su un grafico chiamato audiogramma. La soglia è il livello più basso di intensità a cui si possono sentire i toni puri. Il test ai toni puri comprende anche il test di conduzione per via ossea. Durante questo tipo di test, si indossa una fascia sulla testa con vibratore appoggiato sulla mastoide , mentre sono presentati i toni. È possibile ascoltare i suoni che vengono condotti all’orecchio interno attraverso il cranio. Un audiologo poi confrontare i risultati dei toni puri condotti per via aerea ,con quelli trasmessi per via ossea e determinare quale parte dell’orecchio è responsabile di qualsiasi perdita uditiva.
4. Emissioni Otoacoustiche (OAE)
La coclea è la parte dell’orecchio che riceve informazioni dai suoni in entrata e invia i suoni al cervello. La coclea contiene molte terminazioni nervose chiamate “cellule ciliate”. Quando questi terminazioni nervose lavorano correttamente, i suoni vengono inviati al cervello in maniera accurata ed il cervello interpreta questi suoni come la musica, la parola, o il rumore. Le coclee normali hanno le cellule ciliate esterne che producono rumore. Il test’s OAE registra i suoni che l’orecchio produce e aiuta l’audiologo a determinare se il vostro orecchio interno trasmette il suono al cervello.
5. Auditory Brainstem Response (ABR)
Il test ABR è uno strumento diagnostico non invasivo che misura le onde cerebrali in risposta ad uno stimolo uditivo. Esso misura l’attività della via uditiva che si trova nel vostro tronco encefaloo Per ulteriori informazioni sui test ABR, andare a questi siti.:
Le prove di cui sopra sono solo alcuni dei possibili modi in cui gli audiologi determinano se l’udito è normale. Per saperne di più sulla diagnosi della perdita dell’udito, visitare i seguenti siti web:
American Speech and Hearing Association
Le opzioni per il trattamento WS variano a seconda delle particolari caratteristiche WS. Parte di udito trattamento di perdita è l’intervento di un consulente genetico. Il trattamento può includere uno o una combinazione delle seguenti opzioni. Ricordate che il trattamento (s) scelto da una persona con WS, non può essere adatto per voi o il vostro bambino.
1. Genetic Testing & Counseling
2. Opzioni per il trattamento di perdita di udito includono:
- Apparecchi acustici o Amplification
- Impianti cocleari
- Sign Language
- Logoterapia
Voi o il vostro bambino può scegliere tra una varietà di scuole per sentire gli individui con problemi. Considerate le esigenze specifiche del vostro bambino e il grado di perdita di udito quando si sceglie una scuola. Molte scuole specializzate in formazione per le persone con problemi di udito. Alcune scuole si affidano completamente sulla lingua dei segni americana, alcuni includono una combinazione del linguaggio parlato e segni orale, e altri usano il discorso solo orale. Oltre alle scuole di specializzazione, si può scegliere di inviare il vostro bambino a una scuola pubblica all’interno della vostra comunità. ‘Gli Stati Uniti individui con disabilità Education Act (IDEA) afferma che “ogni bambino ha diritto a una istruzione gratuita e adeguata nel contesto meno restrittivo.” In altre parole, il bambino deve essere dotato delle risorse adeguate in una scuola vicino te.
Caratteristiche fisiche
Caratteristiche fisiche dei WS sono classificati come maggiore o minore. Per essere diagnosticati con WS una persona deve dimostrare due caratteristiche principali, o una maggiore e due minori caratteristiche della sindrome. Non tutti coloro che hanno WS avrà tutte queste caratteristiche, e non tutte le caratteristiche della sindrome sarà presente alla nascita. Alcune caratteristiche si sviluppano più tardi nella vita, e alcuni diventano più pronunciati con l’età.
Le principali caratteristiche sono elencate di seguito. Le descrizioni di ciascuna si trovano qui .
• Eterocromia Iridis
• luminosi occhi azzurri
• Distopia dei canti (spostamento laterale angoli interni palpebrali )
• Broad, radice del naso prominente
• Piccolo mid-face
• capelli prematuramente grigi
• sordità neurosensoriale congenita
• WS Tipo 4 & Malattia di Hirschsprung
Eterocromia Iridis
|
|
|


Occhi molto chiari o brillanti azzurri, occhi di due colori diversi (eterocromia completa), o gli occhi con un’ iride con due colori diversi (eterocromia settoriale);”Eterocromia Iridis” significa che qualcuno, o un animale, ha gli occhi di due colori diversi, come fa il gatto nella foto a destra. Può anche significare due colori nella stessa dell’occhio, come nella foto a sinistra. Questa persona è nata con gli occhi azzurri, e sviluppato macchie di marrone durante l’adolescenza.
Eterocromia rende occhi sguardo insolito, ma non influenza la visione della persona. WS solito non causare problemi di visione.
È interessante notare che gli animali, tra cui cavalli, cani, topi e gatti possono avere WS. Animali sordi fanno i buoni animali domestici, ma sono spesso distrutti nella convinzione che essi non possono essere addestrati. Animali sordi possono fare buoni animali domestici, se sono tenuti in un ambiente protetto.
|
|
|


Le persone con WS hanno spesso occhi blu brillanti belli, in combinazione con una o più delle altre caratteristiche di WS. Occhi azzurri e prematuramente capelli grigi si verificano spesso in combinazione, come fanno nella foto a sinistra. Qualsiasi persona con gli occhi azzurri può avvertire fotofobia, difficoltà di vedere alla luce del sole, perché le persone con gli occhi azzurri non hanno lo strato sovrastante di pigmento scuro che le persone con marrone, nocciola, o gli occhi verdi fanno. Questo pigmento aggiuntivo protegge gli occhi dalla luce forte. Le persone con gli occhi azzurri anche spesso hanno scarsa visione notturna. Gli occhi azzurri da soli non sono diagnostici di WS; tuttavia, gli occhi azzurri in combinazione con i capelli precocemente grigi, aree di pelle depigmentazione, perdita dell’udito suggeriscono WS.
|
|
|


La “Distopia dei canti” descrive l’aspetto degli occhi della persona e ponte nasale. Le persone con distopia dei canti hanno ampie, ponti nasali piatte, con pieghe di pelle che copre angoli interni degli occhi. Queste pieghe della pelle danno un aspetto asiatico agli occhi di persone con WS. Persone asiatici come quello nella foto a destra, hanno pieghe cutanee copre gli angoli interni degli occhi chiamato epicanto. Queste pieghe sono una caratteristica del viso normale causata dai ponti nasali pianeggianti tipici di persone asiatiche. Dystopia dei canti, come mostrato nella foto a fianco, non è una condizione normale per persone di ogni razza. I medici utilizzano una formula denominata “W Index” per calcolare la distanza tra gli occhi per stabilire se qualcuno ha distopia dei canti.
Broad nasale Root
 Broad, importanti radici nasali si trovano spesso nelle persone con WS. La radice nasale è l’area tra gli occhi del ponte della occhiali poggia. Le persone con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e caratteristici nevi di WS.
Broad, importanti radici nasali si trovano spesso nelle persone con WS. La radice nasale è l’area tra gli occhi del ponte della occhiali poggia. Le persone con importanti radici nasali sembrano avere piani, facce larghe, ei loro occhi possono apparire da impostare ampiamente a parte. La persona nella foto ha anche le brillanti occhi azzurri, volumi bruciati sopracciglia, piccolo alae nasale, e caratteristici nevi di WS.
Piccola Mid-Face
 Le persone con WS possono avere ossa facciali che sono più piccoli del normale. Queste ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le occhiaie. Quando ossa metà viso sono più piccolo del previsto, il viso della persona può apparire piatta nel profilo. Ossa nasali appiattite possono causare gli occhi della persona a comparire ampiamente impostare e “orientale-looking”, anche se la persona non è asiatico.
Le persone con WS possono avere ossa facciali che sono più piccoli del normale. Queste ossa sono le ossa nasali, guancia ossa, e alcune ossa che formano le occhiaie. Quando ossa metà viso sono più piccolo del previsto, il viso della persona può apparire piatta nel profilo. Ossa nasali appiattite possono causare gli occhi della persona a comparire ampiamente impostare e “orientale-looking”, anche se la persona non è asiatico.
Quest’uomo ha piccole ossa della faccia media, eterocromia dell’iridis e capelli precocemente grigi – tutte le caratteristiche di WS.
Capelli prematuramente grigi, o macchie di capelli grigi nei capelli di colore più scuro sono anche segni di WS. Molte persone con WS hanno una striscia bianca di capelli al centro della loro fronte. Altri possono avere i capelli che si è interamente grigio quando erano adolescenti. Alcune persone con WS hanno ciglia e sopracciglia bianche. I capelli grigi si verifica spesso in combinazione con gli occhi blu e la perdita dell’udito. Persone i cui capelli sono diventati grigi prematuramente possono scegliere di tingersi i capelli, quindi questa funzione non è sempre visibile in qualcuno con WS. L’uomo nella foto (in alto a destra) ha i capelli che sono diventati grigi quando era adolescente.
Sordità neurosensoriale
 Ipoacusia neurosensoriale può essere presente alla nascita (congenita), o può apparire più tardi nella vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita dell’udito in un solo orecchio, e alcuni individui hanno perdite uditive molto lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell’udito non è una caratteristica visibile, le persone che hanno alcune delle altre caratteristiche di WS devono sempre ricevere una valutazione dell’udito. Un ragazzo con un impianto cocleare è raffigurato qui sotto.
Ipoacusia neurosensoriale può essere presente alla nascita (congenita), o può apparire più tardi nella vita. Nei casi più gravi soggetti con WS sono nati sordi. Alcuni individui hanno la perdita dell’udito in un solo orecchio, e alcuni individui hanno perdite uditive molto lievi, che non interferiscono con la parola o lo sviluppo del linguaggio. Poiché la perdita dell’udito non è una caratteristica visibile, le persone che hanno alcune delle altre caratteristiche di WS devono sempre ricevere una valutazione dell’udito. Un ragazzo con un impianto cocleare è raffigurato qui sotto.
Caratteristiche minori di WS
Caratteristiche di minore importanza sono i seguenti, con le descrizioni disponibili qui :
• leucoderma congenita
• Diverse nevi (moli)
• anomalie sopracciglio
• ipoplasico nasale alae
• Sindattilia
• mento sfuggente
• labio-palatoschisi
• Spina bifida
• Disturbi della comunicazione
• Camptodattilia
• caratteristiche rare

Le persone con WS volte hanno “leucoderma congenita”, o l’assenza di pigmento nella loro pelle. Perché queste macchie bianche o chiare di pelle non hanno il pigmento, si scottature facilmente e sono a rischio di sviluppare il cancro della pelle. Leucoderma è facilmente visibile sullepersone con la pelle scura, e può causare problemi estetici o sociali per queste persone. Ecco una foto di leucoderma su una mano.

Nei Multipli
I nei sono raccolte di cellule pigmentate. I nei sono spesso dispersi attraverso la pelle di persone con WS, solitamente nelle zone della testa e del collo, come nella donna in questa fotografia. I nei dovreb-
bero essere controllati, per essere certi che non diventino maligni, e se necessario dovrebbe essere rimosso chirurgicamente.

Eyebrow Anomalie
Anomalie del sopracciglio sono comuni nelle persone con WS. Le sopracciglia possono essere depigmentate e possono crescere attraverso il ponte del naso e riunite sulla linea mediana. Questa condizione si chiama “synophrys,” o unibrow. Sopracciglia folte o svasate possono creare un viso con aspetto insolito. Nella foto a sinistra,vengono visualizzati sul ragazzo Sopracciglia svasatE
 “Ipoplasia” indica sottosviluppati. Le persone con WS possono avere ipoplasia dei muscoli della spalla e del collo, così come sottosviluppo delle ossa della faccia media. Sono comuni anche ali nasali ipoplasiche (ai lati della punta del naso) nelle persone con WS.
“Ipoplasia” indica sottosviluppati. Le persone con WS possono avere ipoplasia dei muscoli della spalla e del collo, così come sottosviluppo delle ossa della faccia media. Sono comuni anche ali nasali ipoplasiche (ai lati della punta del naso) nelle persone con WS.
Questa donna ha ali nasali ipoplasiche ed ipoplasia dei muscoli della spalla, così come suo figlio. Ha anche eterocromia iridis e nevi multipla, visibile in questa fotografia.
Sindattilia
 Le persone con le dita dei piedi fusi hanno una condizione chiamata “sindattilia.” Sindattilia e polidattilia (troppe dita delle mani o dei piedi) si trovano talvolta nelle persone con WS. Dita fuse sono raffigurati a sinistra.
Le persone con le dita dei piedi fusi hanno una condizione chiamata “sindattilia.” Sindattilia e polidattilia (troppe dita delle mani o dei piedi) si trovano talvolta nelle persone con WS. Dita fuse sono raffigurati a sinistra.

Stempiato Chin
“Mandibola retrognatica” è un altro nome per una piccola mascella inferiore, o mento sfuggente. Le persone con WS possono avere retrognazia nonché orecchie inclinate con inclinazione verso la parte posteriore della testa. La mascella di questa donna è retrognatica (foto a destra). È anche possibile vedere nevi vicino al naso e sul collo.
Labbro leporino e palatoschisi
 La palatoschisi “accade quando il tetto della bocca non forma correttamente in utero. Un palato che non si è fuso è chiamato palatoschisi. Un labbro che non si è fuso è chiamato labbro leporino. Labio e / o palatoschisi sono talvolta presente nei neonati che hanno anche WS. Queste condizioni devono essere corrette chirurgicamente in modo che la persona possa imparare a mangiare e parlare normalmente.
La palatoschisi “accade quando il tetto della bocca non forma correttamente in utero. Un palato che non si è fuso è chiamato palatoschisi. Un labbro che non si è fuso è chiamato labbro leporino. Labio e / o palatoschisi sono talvolta presente nei neonati che hanno anche WS. Queste condizioni devono essere corrette chirurgicamente in modo che la persona possa imparare a mangiare e parlare normalmente.
Questa donna è nata con il labbro leporino e palatoschisi e ora ha un volto normale ricerca e ottime capacità di linguaggio.
Per ulteriori informazioni, consultare il americana palatoschisi – cranio Association (ACPCA) sito web.
“Spina bifida,” o della colonna vertebrale aperto, a volte si verifica nei bambini che hanno WS. Spina bifida si verifica quando le ossa della colonna vertebrale non si fondono durante lo sviluppo fetale. Il fluido cerebrospinale (CSF)che è contenuto del midollo spinale può essere spinti attraverso l’apertura verso l’esterno del corpo. Ciò può verificarsi in qualsiasi punto lungo la lunghezza della colonna vertebrale, dalla base del collo, alla base della colonna vertebrale. La spina bifida deve essere corretto subito dopo la nascita per prevenire l’infezione del midollo spinale e del sistema nervoso.

Persone che hanno WS e perdita di udito, palatoschisi o labbro leporino possono avere difficoltà ad imparare a parlare o usare un linguaggio appropriato. Intervento precoce offre la migliore possibilità per questi bambini di imparare a comunicare in modo efficace. Persone che hanno WS di solito non sono cognitivamente compromesse o ritardati mentali. I loro problemi del linguaggio e, se del caso, il risultato di essere in grado di sentire il discorso normale e il linguaggio, e raramente, dalle labbra e palati schisi non riparati. Alcune persone che hanno WS e sorde, preferiscono far parte della comunità dei sordi e imparare a comunicare attraverso il linguaggio dei segni. Altre persone non udenti scelgono di usare apparecchi acustici o impianti cocleari per consentire loro di imparare la lingua e la parola orale.

“Camptodattilia” significa diti che sono curvati verso l’alto e permanentemente flesse, come le dita di questo individuo che ha WS (a destra). La camptodattilia a volte è trattata con la terapia fisica e chirurgia correttiva.
Molto raramente, gli individui con WS possono nascere senza occhi, una condizione chiamata “anoftalmia.” Questo è più probabile che accada se i genitori del bambino erano stretti consanguinei (accoppiamento tra consanguinei). Alcuni bambini nati da accoppiamenti tra consanguinei sono nati con WS e difetti degli arti. Questo è molto rara combinazione di caratteristiche fisiche. Il ritardo mentale è stata osservata in questi bambini, ma è incerto se i problemi cognitivi derivano da WS o da qualche altra causa.
Molte condizioni hanno sintomi simili, e WS non fa eccezione. L’unico modo assolutamente preciso per sapere se qualcuno ha WS è di test genetici. Tuttavia, i test genetici non è ancora disponibile per tutti i tipi di WS. Se pensate che voi o qualcuno che conosci ha WS, assicuratevi di cercare un modello di anomalie, non solo una o due cose.
Alcune altre condizioni hanno caratteristiche che si trovano anche in WS. Queste condizioni di solito causano problemi di pigmento nei capelli, pelle e / o degli occhi. “L’albinismo” si intende la mancanza di pigmento nella pelle, capelli o gli occhi. Un certo numero di problemi fisici causano l’albinismo, alcuni sono ereditarie (genetiche), e alcuni non lo sono. Le descrizioni delle condizioni elencate di seguito sono qui .
· Piebaldism
· Oculocutaneous Albinismo / Oculare
· Vitiligine
· Sindrome di Horner
· Trauma Eye
“Piebaldismo” è una condizione autosomica dominante che è stata fatta risalire a una mutazione del gene specifico. Le persone che hanno piebaldismo sono molto simili a chi ha l’WS. Essi possono avere zone di pelle depigmentazione sulla loro testa e del tronco, nonché sulla loro sopracciglia, palpebre, ciglia e capelli. Essi possono anche avere occhi di due colori diversi (Eterocromia Iridis). La sordità non è caratteristica di piebaldismo, né ampia radice del naso o distopia dei canti. Piebaldismo si trova spesso nei pazienti di origine africana. E ‘impossibile capire la differenza tra piebaldismo e WS solo guardando l’aspetto fisico di qualcuno. Conoscere la storia della perdita di udito famiglia può essere utile nel fare la distinzione, ma l’analisi genetica è l’unico modo di distinguere tra le due condizioni.
Albinismo Oculocutaneo / Oculare
Le persone con “albinismo oculocutaneo” possono avere totale assenza di pigmento nella loro pelle, capelli e occhi. A volte il pigmento manca solo dai loro occhi. In questo caso la persona ha una condizione chiamata “albinismo oculare.” Il paziente può avere fotofobia, strabismo e riduzione del visus. Alcuni pazienti hanno anche nistagmo congenito (oscillazione involontaria dei bulbi oculari). La maggior parte delle persone affette da albinismo oculare hanno una condizione genetica legata al cromosoma X denominata Nettleship-Falls albinismo oculare. Ciò significa che il gene che causa albinismo oculare che si trova sul cromosoma X e viene trasmesso dalla madre al figlio. La forma autosomica recessiva meno comune dell’albinismo oculare è una specifica mutazione genetica e colpisce maschi e femmine in ugual misura.
Le persone con albinismo oculocutaneo tipo 2 sono simili a persone con albinismo oculocutaneo, salvo che la loro pelle e capelli hanno una diminuzione generalizzata di pigmento. Le persone con questo problema hanno la pelle pallida e capelli colorati giallastro. L’Albinismo oculocutaneo può essere presente in WS2, ma sono necessario i test genetici per una diagnosi differenziale accurata, perché l’albinismo oculocutaneo avviene in molte altre sindromi.
L’Organizzazione Nazionale di albinismo e Ipopigmentazione (NOAH) fornisce informazioni e sostegno alle persone che hanno condizioni di pigmentazione. Possono essere contattati al http://www.albinism.org , o PO Box 959 East Hampstead, NH 03.826-0.959.
La “Vitiligine” è una malattia autoimmune causata da mutazioni genetiche sui cromosomi 1, 7, 8 e 4 persone con vitiligine hanno chiazze di depigmentazione sulla pelle, sul viso, mani, piedi, gomiti, ginocchia e torace. I loro capelli possono perdere il pigmento, e le aree depigmentate della pelle possono aumentare di dimensioni. La perdita dell’udito e anomalie cranio-facciali non sono associati con vitiligine. Alcuni casi di vitiligine sembrano essere ereditarie, mentre altri sono associati a trauma, ipertiroidismo, diabete e problemi surrenali. I principali problemi connessi con vitiligine sono cosmetici, specialmente quando questa condizione si verifica in individui con pelle molto scura. A volte questa condizione va via senza trattamento. Per informazioni sui servizi di assistenza e informazioni sulla vitiligine vedere la risorsa seguente:
Storia
Caratteristiche morfologiche tipiche della sindrome di Waardenburg possono essere riconosciuti immediatamente o subito dopo la nascita. Caratteristiche includono tipicamente ciuffo bianco, ampio radice del naso, e iridi ipopigmentate.
I genitori notano che il bambino non reagisce ai suoni.
Esame Fisico
Non tutti i casi esprime tutte le manifestazioni cliniche della sindrome Waardenburg completa e le condizioni di forme fruste sono comunemente osservati. Sulla base di criteri clinici e genetici, sono riconosciute 4 tipi di sindrome di Waardenburg. Tutte le forme mostrano una marcata variabilità, anche all’interno delle famiglie.
|
|
Contrassegnata asimmetria facciale, lagoftalmo, un angolo cadenti destra della foce. Immagine per gentile concessione di Dourmishev LA et al, Cutis 1999 |
|
|
Il viso nel profilo dimostra mancanza dell’ angolo nasofrontale, ipertricosi delle sopracciglia, punta del naso all’insù, e labbro superiore accorciato con l’arco di Cupido pronunciato. Immagine per gentile concessione di Dourmishev LA |
|
|
Fratello e sorella con sindrome di Waardenburg. Immagine per gentile concessione di Dourmishev LA et al, Cutis 1999; 63:139-40 |



Tipo I Waardenburg
 Il Tipo I Waardenburg è il tipo più comune ed è causata da una mutazione nel gene PAX3. Il gene PAX3 è parte di una famiglia di geni che è importante per lo sviluppo di tessuti e organi. Questo gene produce una proteina che controlla altri geni responsabili dello sviluppo di cellule specifiche e parti del corpo, quali ossa facciali e melanociti (cellule che compongono il pigmento melanina) manifestazioni cliniche (ad esempio, ciuffo bianco, macchie della pelle) sono più frequenti nel I tipo.
Il Tipo I Waardenburg è il tipo più comune ed è causata da una mutazione nel gene PAX3. Il gene PAX3 è parte di una famiglia di geni che è importante per lo sviluppo di tessuti e organi. Questo gene produce una proteina che controlla altri geni responsabili dello sviluppo di cellule specifiche e parti del corpo, quali ossa facciali e melanociti (cellule che compongono il pigmento melanina) manifestazioni cliniche (ad esempio, ciuffo bianco, macchie della pelle) sono più frequenti nel I tipo.
La melanina è responsabile per il colore del nostro occhi , capelli e pelle. Sono necessari anche melanociti nel nell’orecchio interno per l’udito. Pertanto, una mutazione nel gene PAX3 significa che le cellule pigmento melanina non possono svilupparsi adeguatamente, portando a vari problemi. Questi problemi comprendono chiazze di capelli bianchi, e le patch o aree di pelle che mancano pigmentazione. Gli occhi possono anche avere pigmentazione irregolare. Mancanza di melanina contribuisce anche alla neurosensoriale sordità associata alla sindrome. L’importo della perdita dell’udito può essere ovunque da moderata a profonda.
Il Tipo I Waardenburg è generalmente autosomica dominante – è ereditato da un solo genitore.
Tipo II Waardenburg
Waardenburg tipo II, anche un tipo più comune, perdita dell’udito neurosensoriale (77%) e eterocromia iride (47%) sono i due più importanti indicatori diagnostici di questo tipo. si possono anche avere cambiamenti di colore nei capelli, pelle . La differenza principale tra tipo I e tipo II è che nel tipo II, gli occhi non sono distanziati . Mentre si sospetta che questo tipo sia causato da mutazioni nel gene PAX3, si è anche creduto sia causata da mutazioni nel MITF (fattore di trascrizione associato a microftalmia-) ed ai geni SNAI2. Tipo II può essere ereditata sia nella modalità autosomica dominante (un genitore) che con modalità autosomica recessiva, il che significa che due genitori devono avere il gene per essere in grado di produrre un bambino con Waardenburg di tipo II.
Nei tipi autosomici dominanti, un bambino deve ricevere solo il gene WS da un genitore per avere WS. Nei tipi autosomici recessivi, un bambino deve ricevere il gene WS da entrambi i genitori per avere WS.
In aggiunta, ci sono cinque sottotipi di tipo II Waardenburg. Questi sottotipi sono Tipo IIA, che è causata da una mutazione nel gene MITF sul cromosoma 3; Tipo IIB, che è associato con cromosoma 1; Tipo IIC e IID, che coinvolgono il cromosoma 8; e Digitare IIE, una mutazione sul cromosoma 22.

Sindrome di Waardenburg tipo 2 in una famiglia turca: implicazioni per l’importanza del modello di fondo pigmentazione
Tipo III Waardenburg
Poi c’è il Tipo III, un tipo molto raro. è più rara rispetto alle WS1 e WS2, ma è la
forma più grave. Può essere considerata come un sottotipo della WS1 dal momento che anch’essa è dovuta a mutazioni nel gene PAX3 (delezione eterozigote); di solito è sporadica e solo occasionalmente sono stati segnalati casi familiari ( Ciò che distingue il tipo III (anche causata da una mutazione nel gene PAX3) dagli altri è che nel Tipo III, esistono anomalie degli arti superiori, quali malformazioni delle braccia e delle mani, ma è anche caratterizzato da anomalie muscoloscheletriche (cioè, aplasia delle prime due costole, scapola alata, rigidità delle articolazioni, contratture articolari in flessione delle dita delle mani e dei piedi la mancanza di differenziazione delle piccole ossa carpali, la formazione cistica del sacro, anomalie del armi [ad esempio, amiloplasia e rigidità delle articolazioni). Ad esempio, le dita possono essere fuse insieme sindattilia cutanea bilaterale. Questo tipo di Waardenburg è anche conosciuta come sindrome di Klein-Waardenburg. Il Tipo III può sorgere spontaneamente (nessun legame genetico, si ha solo una mutazione ) oppure può essere ereditata da un genitore. o Altre manifestazioni cliniche della sindrome tipo III comprendono la piena sintomatologia della malattia, più ritardo mentale, microcefalia, e gravi anomalie scheletriche.

Figura 1: (a) Sindattilia delle dita medio e anulare delle mani e le dita dei piedi 2 ° e 3 ° di metri lungo con un forte emarginati, macule depigmentate sulla tibia destra. (B) Sindattilia delle dita dei piedi 2 ° e 3 ° del piede sinistro. (C) Sindattilia delle dita dei piedi 2 ° e 3 ° dei piedi giusti. (D) Close-up che mostra 2 ° e 3 ° fuso dita dei piedi
La WS3 è più spesso associata a perdita progressiva dell’udito neurosensoriale.
In un caso insolito, specialisti in India hanno diagnosticato una Waardenburg di tipo III in un bambino di 7 anni in cui si erano fusi il dito medio ed anulare su entrambe le mani, più due dita fuse. Una gamba aveva un cerotto bianco. I suoi occhi erano di colori diversi. Aveva una grande radice del naso (la zona tra gli occhi) e qualche capello bianco. Il padre del bambino era normale e non aveva sintomi. Ma la madre ha avuto sintomi, tra cui la sordità. In seguito è stata diagnosticata come tipo II.
Non è possibile capire la differenza tra WS di Tipo 1 e WS di Tipo 3 basandosi solo sull’aspetto fisico. I test genetici sono l’unico metodo accurato per distinguere tra i due tipi.
Tipo IV Waardenburg
L’ultimo tipo di sindrome di Waardenburg, anche rare, è di tipo IV. il tipo IV è un tipo II con malattia di Hirschsprung. Attualmente, sono stati identificati sei geni, onnicomprensivi nella Tabella 1. Questo tipo è anche conosciuta come sindrome di Waardenburg-Shah (associazione di sindrome di Waardenburg con la malattia di Hirschsprung).Il tipo 4 è raro, con solo 48 casi segnalati fino al 20026 Egbalian F (2008).
Oltre alle caratteristiche tipiche di Waardenburg, questo modulo comprende anche la malattia di Hirschsprung, un problema digestivo che causa stitichezza o blocco intestinale. Il tipo IV viene solitamente tramandato da un genitore, ma può anche essere ereditata da entrambi i genitori.
Le persone con WS4 possono anche avere una condizione chiamata malattia di Hirschsprung (HD). HD di solito viene scoperto poco dopo la nascita, e può essere in pericolo di vita. È una condizione che comporta problemi del colon e l’incapacità di espellere feci dall’intestino. Qualcuno con HD non trattati sviluppa un colon allargata, stipsi cronica, e ritardo di crescita. HD è trattata con la chirurgia correttiva o modificazione della dieta.
Il tipo IV dispone anche di sottotipi, ognuno causata da una mutazione del gene diverso.
La sindrome di Waardenburg-Shah (WSS) è una neurocristopatia caratterizzata dall’associazione tra la sindrome di Waardenburg (sordità neurosensoriale e anomalie della pigmentazione; si veda questo termine) e la malattia di Hirschsprung (si veda questo termine). La prevalenza è sconosciuta ma sono stati riportati oltre 50 casi. I pazienti, nel periodo neonatale, presentano anomalie della pigmentazione (frezza sulla fronte, sopracciglia e ciglia bianche, eterocromia dell’iride, macchie cutanee bianche) in associazione a ostruzione intestinale. La sordità neurosensoriale è comune, precoce e può essere unilaterale. Lo sviluppo psicomotorio è normale. La WSS è causata da anomalie della migrazione/differenziazione delle cellule della cresta neurale durante lo sviluppo embrionale. Fino ad ora sono stati identificati tre geni-malattia: il gene EDNRB (13q22.3), che codifica il recettore dell’endotelina-B, il gene EDN3 (20q13.32), che codifica il ligando del recettore dell’endotelina e il gene SOX10 (22q13.1), che codifica il fattore di trascrizione SOX10. Le mutazioni dei geni EDNRB e EDN3 si trasmettono con modalità autosomica recessiva; i soggetti omozigoti per le mutazioni in questi geni presentano la WSS, mentre quelli eterozigoti presentano la malattia di Hirschsprung isolata o, spesso, sono asintomatici. Le mutazioni del gene SOX10 vengono ereditate con modalità autosomica dominante e alcune specifiche mutazioni (in particolare quelle che interessano i due ultimi esoni codificanti) si associano a una forma più grave della WSS, con quadri neurologici caratterizzati da neuropatia periferica demielinizzante, leucodistrofia centrale dismielinizzante, sindrome di Waardenburg e malattia di Hirschsprung (PCWH; si veda questo termine). Viceversa, alcuni soggetti con la sindrome di Waardenburg tipo 2 (si veda questo termine) sono portatori delle mutazioni eterozigoti del gene SOX10. Non sono state riscontrate mutazioni nei tre geni-malattia nel 20-40% dei soggetti affetti da WSS/PCWH. La diagnosi si basa sul riconoscimento del quadro clinico e viene confermata dal riscontro di mutazioni in uno dei geni-malattia. La diagnosi differenziale si pone con altre forme della sindrome di Waardenburg (1, 2 e 3; si vedano questi termini) e anche con altre malattie rare, come il piebaldismo e le sindromi ABCD o BADS (sempre associate a mutazioni del gene EDNRB; si vedano questi termini). Alle famiglie nelle quali viene riscontrata la mutazione patogenetica deve essere offerta la diagnosi prenatale molecolare. La consulenza genetica deve adattarsi alla modalità di trasmissione della mutazione identificata. Il trattamento è solo sintomatico; il trattamento della malattia di Hirschsprung deve comprendere anche la chirurgia e la sordità deve essere trattata. La prognosi è spesso buona, ma la sindrome può associarsi a elevata morbilità e mortalità, secondarie alle complicazioni della malattia di Hirschsprung (legate alle dimensioni del segmento di intestino aganglionico).
Diagnosi
I bambini nati con la sindrome di Waardenburg possono avere i cambiamenti caratteristici dei capelli e della pelle e perdita dell’udito. Tuttavia, se i sintomi sono lievi, la sindrome Waardenburg può non essere diagnosticata fino a quando non viene diagnosticata un membro della famiglia ed allora tutti i membri della famiglia sono esaminati. Test dell’udito formali possono essere utilizzati per rilevare la perdita dell’udito.
Trattamento
Diversi i sintomi della sindrome di Waardenburg appaiono in persone diverse, anche all’interno della stessa famiglia. Alcuni individui non richiedono alcun trattamento, mentre altri possono avere bisogno di un intervento chirurgico o degli occhi o altre anomalie. Non sono necessari dieta o attività particolari restrizioni, e la sindrome di Waardenburg di solito non influenzano la mente.
La consulenza genetica
A causa del modo con cui la sindrome di Waardenburg (autosomica dominante) di tipo 1 e 2 è ereditata, un individuo affetto ha una probabilità del 50%, ad ogni gravidanza, di avere un figlio affetto. Dal momento che i sintomi possono variare, non c’è modo di prevedere se un bambino affetto avrà sintomi più lievi o più gravi rispetto la sua / il suo genitore. Ereditarietà dei tipi 3 e 4 è più complessa, ma la consulenza genetica può aiutare a valutare il rischio di trasmettere la sindrome di Waardenburg a un bambino.
Diagnosi differenziale
· Piebaldismo
· Sindrome di Vogt-Koyanagi-Harada
Studi di laboratorio
Tipi di sindrome di Waardenburg 1 e 3 sono più comunemente associati con mutazioni puntiformi in PAX3, e di tipo 2 è associato con MITF mutazioni puntiformi. Il piebaldismo (OMIM 17200) è una rara patologia autosomico dominante caratterizzata dalla presenza di macchie ipopigmentate
a livello della parte mediana della fronte e diffuse a livello di torace, addome ed arti. È causato da mutazioni
inattivanti o delezioni del gene KIT o del gene SLUG.Legatura-dipendente multipla della sonda amplificazione può essere utilizzato per rilevare cambiamenti nei geni bersaglio Milunsky JM,et al.,2007.
Altri test
Partington ha usato le 3 distanze interoculari per determinare la presenza di distopia cantorum. (Il valore referente tra distanza angoli palpebrali mediale delle palpebre e la lunghezza tra gli alunni era di 0,6 mm.)
· Tra gli angoli cantali interni
· La vicina distanza papillare
· Tra gli altri angoli cantali
Arias e Mota ha sviluppato l’indice W per la diagnosi di spostamento laterale della angoli palpebrali interiore. (Indice AW superiore a 2,07 mostra dystopy, mentre un indice inferiore a 1,87 è normale.)
· W index = X + Y + alb
· Dove X = [a – {0.21119c + 3,909}] / c e Y = [2a – {0.2497b + 3,909}] / b in mm
· Distanza a è tra angoli palpebrali interni; distanza b, tra gli alunni; e la distanza c, tra angoli palpebrali esterno.
I risultati istologici
Gli studi istochimici nella pelle acromica delle persone con sindrome di Waardenburg mostrano che i melanociti sono assenti, o che solo poche cellule diidrossifenilalanina-positive sono presenti.
Osservazioni ultrastrutturali non rivelano melanociti, cellule dendritiche indeterminati, o melanosomi nei cheratinociti della pelle depigmentazione; tuttavia, il numero di cellule di Langerhans nell’epidermide è normale. Il numero di melanociti sul bordo delle leucodermatiti è ridotta, e numerose anomalie nucleari citoplasmatici sono noti. Alcuni melanosomi circondate da un alone chiaro si trovano all’interno dei vacuoli. Il numero dei melanociti nelle zone ipopigmentate della sindrome di Waardenburg è drasticamente ridotto. Queste cellule dendritiche contengono melanosomi mal melanizzate.
Esame istopatologico delle orecchie interne delle persone con sindrome di Waardenburg mostra organi assenti Corti, atrofia del ganglio spinale, e una riduzione del numero di fibre nervose.
Tipi di WS 1 e 3
Tipi di WS 1 e 3 hanno caratteristiche fisiche molto simili. Tipi 1 e 3 hanno un modello di ereditarietà autosomica dominante. Questo significa che se un genitore ha il gene per WS e l’altro genitore non lo fa, i loro figli hanno una probabilità del 50% di avere WS.
WS1 è associato con congeniti, variabili neurosensoriali (permanenti) perdite uditive.
WS3 è più spesso associata a perdita progressiva dell’udito neurosensoriale.
Entrambi i tipi hanno le caratteristiche fisiche descritte altrove su questo sito. Queste funzionalità includono eterocromia, luminosi occhi azzurri, distopia dei canti, e le anomalie del pigmento.
WS3 ha le caratteristiche aggiuntive di anomalie degli arti superiori e minori contratture articolari delle dita delle mani e dei piedi.
Non è possibile capire la differenza tra WS Tipo 1 e Tipo 3 WS basata sull’aspetto fisico. Test genetici è l’unico modo accurato per distinguere tra i due tipi.
Gli individui con WS2 possono avere caratteristiche simili a quelle di WS 1 e 3, tranne che dei canti distopia e anomalie muscoloscheletriche non sono presenti.
WS di tipo 2
- WS2 ha diversi sottotipi in base alla posizione del gene specifico che causa problemi. La maggior parte dei sottotipi sono autosomica dominante in eredità, ma alcuni sono autosomica recessiva. Nei tipi autosomiche dominanti, un bambino deve ricevere solo il gene WS da un genitore per avere WS. Nei tipi autosomiche recessive, un bambino deve ricevere il gene WS da entrambi i genitori per avere WS.
- Le principali caratteristiche del WS2 sono iridis heterochromia e ipoacusia neurosensoriale.
WS Type 4 è anche chiamato sindrome di Shah-Waardenburg.
- Persone con WS4 possono avere uno o tutti i sintomi di WS2.
Le persone con WS4 hanno una condizione chiamata malattia di Hirschsprung (HD). HD di solito viene scoperto poco dopo la nascita, e può essere in pericolo di vita. HD è una condizione che comporta problemi al colon, e l’incapacità di espellere feci dall’intestino. Qualcuno con HD svilupperà un colon allargata, la stipsi cronica, ritardo di crescita, e, se l’intervento chirurgico è riuscito, può morire.
I sottotipi della sindrome sono riconducibili a diverse varianti genetiche:
|
Tipo |
Gene |
Conosciuto anche come |
||
|
Tipo I, WS1 |
2q35 |
– |
||
|
Tipo IIa, WS2A (originariamente WS2) |
3p14.1-p12.3 |
– |
||
|
Tipo IIb, WS2B |
1p21-p13.3 |
– |
||
|
Digitare IIc, WS2C |
8p23 |
– |
||
|
Digitare IId, WS2D (molto raro) |
8q11 |
– |
||
|
Tipo III, WS3 |
2q35 |
Sindrome di Klein-Waardenburg |
||
|
Tipo IVa, WS4A |
13q22 |
|||
|
Tipo IVb, WS4B |
20q13 |
|||
|
Digitare IVc, WS4C |
22q13 |
|
Altri collaboratori
- Sindrome di Waardenburg-Klein prende il nome Petrus Johannes Waardenburg (1886-1979), un oculista olandese e genetista, e David Klein , un genetista umano svizzero e oculista.
- La sindrome di Mende II è il nome di Irmgard Mende (1938 -), un medico tedesco-americano.
- Van der Sindrome Hoeve-Halbertsma-Waardenburg prende il nome da Jan Van der Hoeve (1878-1952), un oculista olandese, Nicolaas Adolf Halbertsma (1889-1968), medico olandese e Petrus Johannes Waardenburg (1886-1979).
- Van der sindrome Hoeve-Halbertsma-Gualdi prende il nome da Jan Van der Hoeve (1878-1952), Nicolaas Adolf Halbertsma (1889-1968) e Vincenzo Gualdi (1891-1976), un medico italiano.
- La sindrome di Vogt è chiamato per Cecile Vogt (1875-1962), un neuropatologa franco-tedesca.
BIBLIOGRAFIA
Arias, S., & Mota, M. (1978). Apparent non-penetrance for dystopia in Waardenburg’s syndrome type 1, with some hints on the diagnosis of dystopia canthorum. J. Genet. Hum., 26 , 103-131.
^ Arias S (1971). “Genetic Heterogeneity in the Waardenburg Syndrome”. Birth Defects Original Article Series 7 (4): 87–101.
Baar-Hamilton, R., Matheson, L., & Keay, D. (1991). Ototoxicity of cis-platinum and its relationship to eye colour. The Journal of Laryntology and Otology, 105, 7-11.
Baldwin, C., Hoth, C., Macina, R., & Milunsky, A. (1995). Mutation in PAX3 that causes Waardenburg’s syndrome type I: Ten new mutations and review of the literature. American Journal of Medical Genetics, 58 : 115-122.
Bandyopadhyay, S., Prasad, S., & Singhania, P. (1999). Partial anodontia in a case of Waardenburg’s syndrome. The Journal of Laryngology and Otology, 113 : 672-674.
^ Baral, Viviane, et al. “Screening of MTIF and SOX10 Regulatory Regions In Waardenburg Syndrome Type 2. “Plos ONE 7.7 (2012): 1-8. Academic Search Premier. Web. 4 Apr. 2014.
Black, F., Pesznecker, S., Allen, K., & Gianna, C. (2001). A vestibular phenotype for Waardenburg syndrome. Otology & Neurotology, 22 : 188-194.
Bondurand N, Pingault V, Goerich DE, et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet. Aug 12 2000;9(13):1907-17. [Medline].
Cagatay, O., Baserer, N., & Tinaz, N. (2000). Audiometric manifestations of Waardenburg’s syndrome. Ear, Nose & Throat Journal.
Chan KK, Wong CK, Lui VC, Tam PK, Sham MH. Analysis of SOX10 mutations identified in Waardenburg-Hirschsprung patients: Differential effects on target gene regulation. J Cell Biochem. Oct 15 2003;90(3):573-85. [Medline].
Chang T, Hashimoto K, Bawle EV. Spontaneous contraction of leukodermic patches in Waardenburg syndrome. J Dermatol. Nov 1993;20(11):707-11. [Medline].
Da-Silva, E., Batista, J., Medeiros, M., & Fonteless, S. (1993). Craniofacial anthropometric studies in Waardenburg’s syndrome type I. Clinical Genetics, 44 : 20-25.
DeStefano AL, Cupples LA, Arnos KS, et al. Correlation between Waardenburg syndrome phenotype and genotype in a population of individuals with identified PAX3 mutations. Hum Genet. May 1998;102(5):499-506. [Medline].
Dennis, L., Lanza, J., & Har-el, G. (1996). Waardenburg Syndrome. Otoltolaryngol Head Neck Surgery, 114 : 166-167.
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol. Sep 1999;38(9):656-63. [Medline].
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg’s syndrome with facial palsy and lingua plicata: is that a new type of disease?. Cutis. Mar 1999;63(3):139-41. [Medline].
Dundar M, Lowther G, Colgan J, et al. A case with Waardenburg syndrome presenting with two separate translocations–one reciprocal and one complex. Clin Dysmorphol. Jan 2001;10(1):65-6. [Medline].
^ Egbalian F (2008). “Waardenburg-Shah Syndrome; A Case Report and Review of the Literature” . Iranian Journal of Pediatrics 18(1): 71–74.
Fisch, L. (1959). Deafness as part of an hereditary syndrome. The Journal of Laryntology and Otology , 355-382.
Galasso C, Bombardieri R, Cerminara C, Stranci G, Curatolo P. Anophthalmia-Waardenburg syndrome with expanding phenotype: does neural crest play a role?. J Child Neurol. Nov 2007;22(11):1252-5.[Medline].
^ Geigy CA, Heid S, Steffen F, Danielson K, Jaggy A, Gaillard C (2007). “Does a pleiotropic gene explain deafness and blue irises in white cats?”. Veterinary Journal 173 (3): 548–553.
Graned, G., Collet, L., & Morgon, A. (1995). Physiopathological investigations in a family with a history of Unilateral hereditary deafness. Acta Otolaryngology (Stockh), 115 : 196-201.
Gupta, V., Harisch M., & Aggarwal, C. (2000). Open angle Glaucoma as a manifestation of Waardenburg’s syndrome. Indian J Opthalmol, 48 : 49-50.
Hildesheimer M., Maayan Z., Muchnik C., Rubinstein M., Goodman R.M. Auditory and vestibular findings in Waardenburg’s type II syndrome J. Laryngol. Otol. 1989 ; 103 : 1130-1133
Jung HJ, Jin SA, Choi SJ, Lee SC, Yun SJ. A de novo SOX10 mutation in apatient with Waardenburg syndrome type IV. J Am Acad Dermatol. 2013;68(6):[Medline].
Kadoi, C., Hayasaka, S., & Yamamoto, S. (1996). Branch retinal vein occlusion in a patient with Waardenburg Syndrome. Ophthalmologica, 210 : 354-357.
Keefe, M. (1999). A typical presentation of Waardenburg’s syndrome. Otolaryngol Head Neck Surgery, 120: 749.
Keats, J. (2002). Genes and syndromic hearing loss. Journal Of Communication, 35: 355-366.
Klein, D. (1983). Historical background and evidence for dominant inheritance of the Klein-Warrensburg syndrome(type III). American Journal of Medical Genetics, 14: 231-239.
^Klein-Waardenburg syndrome at Who Named It?
Knox, G., Mcpherson, A., & Lopes, A. (1999). A clinical presentation of Waardenburg’s syndrome type II .
^ Kumar, Sudesh, and Kiran Rao. “Waardenburg Syndrome: A Rare Genetic Disorder, A Report of Two Cases.” Indian Journal of Human Genetics 18.2 (2012): 254-255. Academic Search Premier. Web. 4 Apr. 2014.,
Lee, D., Lanza, J., & Har-el, G. (1996). Waardenburg Syndrome. Otolaryngol Head Neck Surgery , 114: 166-167.
Liu, X., Newton, E., & Read, A. (1995). Hearing loss and pigmentary disturbances in Waardenburg syndrome with reference to WS Type II . The Journal of Laryntology and Otology , 109: 96-100.
Liu, X., Newton, E., & Read, A. (1995). Waardenburg’s syndrome type II: Phenotype findings and diagnostic criteria. American Journal of Medical Genetics, 55: 95-100.
Liu, X. & Newton, E. (1997). Distortion product emissions in normal-hearing and low-frequency hearing loss carriers of genes for Waardenburg’s syndrome. Ann Otol Rhinol Larygology, 106: 220-225.
^ Matulich E. “Deafness in Ferrets” . Cypresskeep.com.
Mckenna, M., Milunsky, A., Baldwin, C., & Nadol, J. (2001). Otopathology in a case of type 1 Waardenburg’s syndrome. Ann Otol Rhinol Otolaryngology, 110.
Madden C, Halsted MJ, Hopkin RJ, Choo DI, Benton C, Greinwald JH Jr. Temporal bone abnormalities associated with hearing loss in Waardenburg syndrome. Laryngoscope. Nov 2003;113(11):2035-41.[Medline].
^ Matulich E. “Deafness in Ferrets” . Cypresskeep.com.
Milunsky JM, Maher TA, Ito M, Milunsky A. The value of MLPA in Waardenburg syndrome. Genet Test. Summer 2007;11(2):179-82. [Medline].
Mishriki, Y. (2000). Facial clues to an inherited syndrome. Postgraduate Medicine , 106.
Moore SW, Johnson AG. Hirschsprung’s disease: genetic and functional associations of Down’s and Waardenburg syndromes. Semin Pediatr Surg. Aug 1998;7(3):156-61. [Medline].
Morell, R., Friedman.,T., Asher, J., & Robbins, L. (1996). The incidence of deafness is non-randomly distributed among families segregating Waardenburg syndrome type 1 (WS1).
Morell R, Carey ML, Lalwani AK, Friedman TB, Asher JH Jr. Three mutations in the paired homeodomain of PAX3 that cause Waardenburg syndrome type 1. Hum Hered. Jan-Feb 1997;47(1):38-41. [Medline].
Nakashima, S., Sando, I., Takahashi, H., & Hashida, Y. (1992). Temporal bone histopathology findings of Waardenburg’s syndrome: A case report. Laryngoscope, 102: 563-567.
Newton, V. (1990). Hearing loss and Waardenburg’s syndrome: implications for genetic counseling. The Journal of Laryntology and Otology, 104, 97-103.
Ortonne, J. (1988). Piebaldism, Waardenburg’s syndrome, and related disorders. Dermatologic Clinics, 6, 205-216.
Oshimo T, Fukai K, Abe Y, Hozumi Y, Yokoi T, Tanaka A, et al. Pediatric case report: clinical profile of a patient with PCWH withp.Q377X nonsense mutation in the SOX10 gene. J Dermatol. 2012;39(12):1022-5.[Medline].
Oysu C, Baserer N, Tinaz M. Audiometric manifestations of Waardenburg’s syndrome. Ear Nose Throat J. Sep 2000;79(9):704-9. [Medline].
^Petrus Johannes Waardenburg at Who Named It?
Read, A. & Newton, V. (1997). Waardenburg syndrome. Journal of Medical Genetics, 34: 656-665.
Read, A. (2000). Waardenburg syndrome. Genetics in Otorhinolaryngology Adv Otorhinolaryngol, 56: 32-38.
Read, A. (2000). Hereditay deafness: lessons for developmental studies and genetic diagnosis. Eur J Pediatr, 159: 232-235.
Reed, W., Stone, V., Boder, E., & Ziprowski, L. (1967). Pigmentary disorders in association with congenital deafness. Arch Derm, 95, 176-178.
Reynolds, J., et al. (1995). Analysis of variability of clinical manifestations in Waardenburg syndrome. American Journal of Medical Genetics, 57: 540-547.
^ Richards J (1999). ASPCA Complete Guide to Cats: Everything You Need to Know About Choosing and Caring for Your Pet .Chronicle Books. p. 71.
Rosenberg, T., et al. (1987). Chroidereima, congenital deafness and mental retardation in a family with an X chromosomal deletion. Ophthalmic Paediatrics and Genetics, 8: 139-143.
Sayh, B., Akarsu, A., & Altan, S. (1995). Anophthalmos-Syndactyly(Waardenburg) syndrome without oligodactyly of toes. American Journal of Medical Genetics, 58: 18-20.
Sener, R. (1998). Cranial MR imaging findings in Waardenburg syndrome: exophthalmia, and hypothalamic hamartoma. Computerized Medical Imaging and Graphics, 22: 409-411.
Sheffer, R. & Zlotogora, J. (1992). Autosomal dominant inheritance of Klein Waardenburg’s syndrome. American Journal of Medical Genetics , 42: 320-322.
Shim WK, Derieg M, Powell BR, Hsia YE. Near-total intestinal aganglionosis in the Waardenburg-Shah syndrome. J Pediatr Surg. Dec 1999;34(12):1853-5. [Medline].
Smith, S., Kolodziej, P., & Olney, A. (1998). Waardenburg Syndrome. Ear, Nose& Throat Journal, 77:257-258.
Sotirova, V., et al. (1999). Identification of a novel mutation in the paired domain of PAX3 in an Iranian family with Waardenburg’s Sydrome type 1. Opthalmic Genetics , 21:25-28.
Strain, M. et al.(1992). Brainstem auditory-evoked potential assessment of congenital deafness in Dalmatians: Associations with phenotypic markers. Journal of Veterinary Internal Medicine , 6: 175-182.
Sudip Kumar Ghosh, Debabrata Bandyopadhyay, Arghyaprasun Ghosh, Surajit Kumar Biswas, Rajesh Kumar Mandal.Waardenburg syndrome: A report of three cases. Indian Journal of Dermatology, Venereology and Leprology. Vol. 76, Num. 5, 2010, pp. 550-552
Suyugul, Z., et al. (1996). Anophtalamia-Waardenburg Syndrome: A report of three cases. American Journal of Medical Genetics, 62: 391-397.
Sznajer Y, Coldea C, Meire F, Delpierre I, Sekhara T, Touraine RL. A de novo SOX10 mutation causing severe type 4 Waardenburg syndrome without Hirschsprung disease. Am J Med Genet A. Apr 15 2008;146A(8):1038-41. [Medline].
Tachibana M. Evidence to suggest that expression of MITF induces melanocyte differentiation and haploinsufficiency of MITF causes Waardenburg syndrome type 2A. Pigment Cell Res. Feb-Apr 1997;10(1-2):25-33. [Medline].
Tagra S, Talwar AK, Walia RL, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol. Jul-Aug 2006;72(4):326. [Medline].
Tekin M, Bodurtha JN, Nance WE, et al. Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3:a simple variant or a true syndrome? Clinical Genetics 2001;60:301-4
Valenzuela, G., et al. (1995). Waardenburg syndrome and Gastric Stasis in adults.
Verheij JB, Sival DA, van der Hoeven JH, et al. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature. Eur J Paediatr Neurol. Jan 2006;10(1):11-7.[Medline].
^ Yang, T, et al. “Double Heterozygous Mutations Of MTIF and PAX3 Result in Waardenburg Syndrome With Increased Penetrance In Pigmentary defects.” Clinical Genetics 83.1 (2013): 78-62. Academic Search Premier. Web. 3 Apr. 2014.
Yoder, B. & Preyson, R. (2002). Shah-Waardenburg syhndome and dandy-walker formation: an autopsy report. Clinical Neuropathology , 21: 236-240.
^ Jump up to:ab Waardenburg PJ (September 1951). “A New Syndrome Combining Developmental Anomalies of the Eyelids, Eyebrows and Noseroot with Pigmentary Anomalies of the Iris and Head Hair and with Congenital Deafness; Dystopia canthi medialis et punctorum lacrimalium lateroversa, Hyperplasia supercilii medialis et radicis nasi, Heterochromia iridum totalis sive partialis, Albinismus circumscriptus (leucismus, poliosis) et Surditas congenita (surdimutitas) “ . Am. J. Hum. Genet. 3 (3): 195–253.
^ Webb AA, Cullen CC (2010). “Coat color and coat color pattern-related neurologic and neuro-ophthalmic diseases” . The Canadian Veterinary Journal 51 (6): 653–657.
Wildhardt G, Zirn B, Graul-Neumann LM, Wechtenbruch J, Suckfüll M, Buske A, et al. Spectrum of novel mutations found in Waardenburg syndrome types 1 and 2: implications for molecular genetic diagnostics.BMJ Open. 2013;18;3(3):[Medline].
Zhang H, Luo H, Chen H, Mei L, He C, Jiang L, et al. Functionalanalysis of MITF gene mutations associated with Waardenburg syndrome type 2. FEBS. 30;586(23);2012:4126-31. [Medline].
Books
Cremers, WRJ & Smith, RJH (2002). Genetic hearing impairment: It’s clinical presentations (Advances in oto-rhino-laryngology) . New York, NY: S.Karger AG. (ISBN# 3085574495)
Jones, KL & Smith, DW (2006). Smith’s recognizable patterns of human malformation . Philadelphia, PA: Elsevier, Inc.(ISBN# 0721661157)
Kahn, A. (2007). Waardenburg Syndrome. San Diego, CA: Plural Publishing, Inc. (ISBN# 1597560219)
Toriello, HV, Reardon, W. & Gorlin, RJ (2004). Hereditary hearing loss and its syndromes (Oxford monographs on medical genetics) . New York, NY: Oxford University Press, Inc.(ISBN# 019513849X)
Wiedemann, HR & Kunze, J. (1997). Clinical syndromes (3rd ed.) . Italy: Mosby-Wolfe.(ISBN# 0723429502)
Willems, PJ (2004). Genetic hearing loss . New York, NY: Marcel Dekker, Inc. (ISBN# 0824743091)
Sindrome branchiootorenale BOR, Sindrome BOR, Displasia branchiootorenale, Branchio-Otorenale Displasia,Branchio-Otorenale Syndrome, Sindrome Branchio-Oto-Renale, Sindrome di Melnick-Fraser
– trasmissione autosomica dominante Fig.1
– espressività variabile
– sordità neurosensoriale. mista, trasmissiva Fig.2
– boffoni preauricolari Fig.3
– cisti-fistole del collo (branchiali) Fig.4
– malformazione del padiglione auricolare Fig.5
– malformazioni renali (agenesia-ipoplasia)Fig.6
|
|
|
|
|
Fig.1
|
Fig.2 |
Fig. 3 |
|
|
|
|
|
Fig.4 |
Fig.5 |
Fig.6 |






Che cosa è la sindrome Branchio-Oto-Renale?,pag.2
Quali geni sono legati alla sindrome Branchio-Oto-Renale?,pag.3
Come i pazienti ereditano la sindrome Branchio-Oto-Renale ?,pag.3
Manifestazioni otorinolaringoiatriche, ?pag.3
Eziologia e la patogenesi, pag.4
Diagnosi, Criteri principali, Criteri minori,pag.6
APPROFONDIMENTI
Famiglia con sindrome Branchio-Oto-Renale: correlazioni cliniche e genetiche,pag.11
Un caso di Sindrome di Branchio-Oto-Renale,pag.18
La sindrome Branchio-oculo-facciale e le sindromi branchio-otic/branchio-oto-renale sono entità distinte,pag.22
Che cosa è la sindrome Branchio-Oto-Renale?
LA Sindrome Branchio-Oto-Renale (BOR) è una condizione genetica, nota anche come sindrome di Melnick-Fraser, è caratterizzata da un’associazione di: 1) fistole brachiale o cisti lungo i lati del collo corrispondenti alla posizione delle fessure branchiali embriologiche; 2) malformazioni dell’orecchio, che possono includere l’orecchio interno, medio ed esterno; 3) malformazioni renali, che possono variare in gravità da ipoplasia renale alla agenesia con conseguente insufficienza renale. I segni ed i sintomi di questa condizione , tuttavia, possono variare.
“Branchio-” si riferisce al secondo arco branchiale, che è una struttura che negli embrioni dà origine ai tessuti nella parte anteriore e laterale del collo. Nelle persone con sindrome branchiootorenale , sviluppo anormale del secondo arco branchiale possono causare la formazione delle masse al collo chiamato cisti branchiale leporino. In alcune persone, connessioni anormali chiamati modulo fistole passaggi tra queste cisti e la superficie del collo. Fistole può anche svilupparsi tra la pelle del collo e della gola, vicino alle tonsille. Cisti schisi branchiali e fistole possono causare problemi di salute se diventano infette.
“Oto-” si riferisce all’orecchio; la maggior parte delle persone con sindrome branchiootorenale hanno perdite e altre anomalie dell’orecchio dell’udito. La perdita dell’udito è conosciuta come la sordità neurosensoriale se è causato da cambiamenti nella parte interna dell’orecchio e sordità conduttiva se è causato da cambiamenti dell’orecchio medio. La sindrome Branchio-Oto-Renale può anche comportare la perdita che deriva da cambiamenti sia l’orecchio interno e l’orecchio medio, che si chiama perdita uditiva mista dell’udito. Altre anomalie dell’orecchio associate alla sindrome di Branchio-Oto-Renale comprendono malformazioni dell’orecchio interno o l’orecchio medio e orecchio esterno di forma anomala (padiglioni auricolari). Alcune persone colpite hanno anche piccoli fori nella pelle (pit preauricolari) o piccoli lembi di pelle (tag preauricolari) proprio di fronte l’orecchio.
“Renale” si riferisce ai reni; La sindrome Branchio-Oto-Renale provoca alterazioni della struttura e della funzione renale. Queste anomalie vanno da lievi a gravi e possono interessare uno o entrambi i reni. In alcuni casi, la malattia renale allo stadio terminale (ESRD) si sviluppa più tardi nella vita. Questa grave condizione si verifica quando i reni diventano incapaci di filtrare efficacemente i liquidi ei prodotti di scarto dal corpo.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Ereditata in modo autosomico dominante, ogni bambino di un genitore con la sindrome di BOR ha una probabilità del 50% di presentare con la malattia. Si pensa che il 90% dei casi di sindrome BOR sono dovuti a eredità, mentre il rimanente 10% dei casi sono dovuti a mutazioni acquisite. BOR mostra espressività variabile, che rappresentano le differenze nella gravità dei sintomi tra i membri della famiglia. Penetranza, tuttavia, è 100%. Attesa (propensione per le generazioni successive di presentare con la malattia in età più giovane) non si verifica.
Storia
• Nel 1864 Heusinger è stato il primo a riconoscere un’associazione tra branchiale fistole leporino, pozzi preauricolari, e compromissione dell’udito.
• Nel 1975, Melnick et al. e Fraser et al. descritto sindrome BOR come entità specifica con un pattern di ereditarietà autosomica dominante.
Epidemiologia, Quanto è diffusa la sindrome Branchio-Oto-Renale ?
I ricercatori stimano che la sindrome Branchio-Oto-Renale colpisce circa 1 su 40.000 persone.
Quali geni sono legati alla sindrome Branchio-Oto-Renale ?
Le mutazioni in tre geni, EYA1, SIX1 e SIX5, sono noti per causare la sindrome Branchio-Oto-Renale . Circa il 40 per cento delle persone con questa condizione hanno una mutazione nel gene EYA1. Eredità autosomica dominante indica che il gene difettoso responsabile di una malattia si trova in una autosoma , e solo una copia del gene è sufficiente a causare la malattia, quando ereditato da un genitore che ha il disturbo. Mutazioni SIX1 eSIX5 sono cause molto meno comuni della malattia.
Le proteine prodotte dal EYA1, SIX1, ei geni SIX5 svolgono un ruolo importante nello sviluppo prima della nascita. La proteina EYA1 interagisce con diverse altre proteine, tra cui SIX1 e SIX5, per regolare l’attività dei geni coinvolti in molti aspetti dello sviluppo embrionale. La ricerca suggerisce che queste interazioni proteine sono essenziali per la normale formazione di molti organi e tessuti, tra cui il secondo arco branchiale, orecchie e reni. Mutazioni nel EYA1, SIX1, o gene SIX5 spesso interrompono la capacità delle proteine ‘di interagire uno con l’altro e regolare l’attività dei geni. Le conseguenti variazioni genetiche influenzano lo sviluppo di organi e tessuti prima della nascita, che porta gli elementi caratteristici della sindrome Branchio-Oto-Renale .
Mutazioni Eya1 e SIX1 sono stati trovati anche in alcune persone con una condizione nota come branchio-oto (BO) sindrome. Questa condizione include molte delle stesse caratteristiche come la sindrome branchiootorenale, ma individui affetti non hanno renali (renale) anomalie. Le due condizioni sono altrimenti così simili che i ricercatori spesso li considerano insieme (BOR / sindrome BO). Non è chiaro il motivo per cui alcune mutazioni Eya1 e SIX1 sono associati ad anomalie renali e altri non lo sono.
Alcune persone con la sindrome di Branchio-Oto-Renale non hanno una mutazione identificata nel EYA1, SIX1,o gene SIX5. In questi casi, la causa della condizione è sconosciuta.
Per saperne di più sul EYA1 , SIX1 , e SIX5 geni.
Come i pazienti ereditano la sindrome Branchio-Oto-Renale ?
Questa condizione è ereditata come carattere autosomico dominante, il che significa una sola copia del gene alterato in ogni cella è sufficiente a causare il disturbo. In circa il 90 per cento dei casi, una persona affetta eredita la mutazione da un genitore affetto. I casi rimanenti derivano da nuove mutazioni nel gene, e si verificano in persone senza storia di malattia nella loro famiglia.
Manifestazioni otorinolaringoiatriche
Le malformazioni auricolari comprendono pozzi preauricolari , che sono ciechi depressioni capocchia di spillo nella parte superiore del padiglione auricolare dell’orecchio. Essi si osservano in 75-85% dei pazienti. Altre malformazioni dell’orecchio esterno (40-60%) possono includere tag preauricolari, lop o pipistrello orecchie e microtia. I canali uditivi esterni possono essere atresici.
Anomalie dell’orecchio medio comprendono anomalie del ossicini, il nervo facciale, ei canali di Falloppio. La tomografia computerizzata delle ossa temporali può dimostrare coclea ipoplasico e canali semicircolari assenti o ipoplasia. È stata inoltre segnalata Mondini displasia. La perdita dell’udito (75-90%) di solito è stabile e può essere conduttiva, neurosensoriale o mista. Si stima che il 2% dei bambini con sordità profonda hanno la sindrome BOR.
Descritta per la prima nel 1975, da M. Melnick, e nel 1978 da FC Fraser. La sindrome di BOR (nota anche come sindrome di Melnick-Fraser) è l’associazione di malformazioni auricolari, fistole branchiali(63%) della sindrome BOR sono solitamente bilaterale e nella parte inferiore del collo con aperture esterne sul bordo mediale del muscolo sternocleidomastoideo, sordità e anomalie renali. La prevalenza è stimata in 1 caso ogni 40.000 popolazione generale.tole branchiali. Altre manifestazioni associate comprendono l’aplasia o stenosi dei dotti lacrimali (8-9%), palato alto arcuato o spacco, morso profondo, e anomalie del nervo facciale. Alcuni autori considerano la microsomia emifacciale (HFM) per essere una forma grave. Vedere emifacciale microsomia, nella sezione Sindrome di Goldenhar. La sindrome branchio-oto-renale (BOR) associa sordità, fistole branchiali multiple e una malformazione renale. La sua prevalenza è stimata a 1/ 40.000(Fraser F.C.). Le malformazioni renali possono essere notevoli (agenesie oppure ipoplasie maggiori) e portano a volte a un’interruzione della gravidanza. Le malformazioni meno importanti saranno diagnosticate con un’ecografia renale che deve essere richiesta di fronte a una sordità suggestiva di BOR: la sordità si accompagna a malformazioni dell’orecchio esterno (malformazioni del padiglione, aplasia dell’orecchio, encondromi, stenosi dei condotti uditivi) (Fig. 4 ), dell’orecchio medio (esiste una componente di trasmissione all’audiogramma) e dell’orecchio interno (varie malformazioni cocleovestibolari) (Fig. 5 ). Si ritrovano in generale delle fistole preauricolari bilaterali e possibilità di apertura di fistole della seconda fessura branchiale con residui cartilaginei associati suggestivi (Fig. 6 ) In presenza di questi segni. un’ecografia renale può confermare questa sindrome.. In pratica, in presenza di una sordità di percezione o mista associata a una fistola branchiale oppure a malformazioni dell’orecchio esterno, è consigliabile eseguire un’ecografia renale. Tre geni sono stati localizzati e due identificati, EYA1 e SIX1(Abdelhak S., Ruf R.G.). Il gene EYA1 può anche essere responsabile di una sindrome branchio-otologica, molto simile alla BOR ma senza interessamento renale(Vincent C.).
· EYA1 (situato sul cromosoma 8 , [8q13.3]), più di 100 tipi di mutazioni responsabili di BOR, le mutazioni sono divisi in tre gruppi principali (isoforma A, B e C) (presente nel 38% -40,5% dei pazienti)
· Six1 (situato sul cromosoma 14 , [14q23.1]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in 5% dei pazienti),
· SIX5 (situato sul cromosoma 19 , [19q13.32]), almeno quattro tipi di mutazioni responsabili della BOR (rilevato in più dell’1% dei pazienti).
Eziologia e la patogenesi
La sindrome BOR viene ereditata come carattere autosomico dominante con elevata penetranza ed espressione variabile. La patogenesi si presume essere una carenza nella differenziazione del primo e secondo arco branchiale. Le anomalie del sistema renale sono il risultato dell’interazione anomala tra la gemma ureterale e il blastema metanefrico. L’orecchio interno (stria vascularis) ed i glomeruli renali condividono un antigene comune, e più di un gruppo di studio ha descritto i cambiamenti istopatologici su esami dell’osso temporale.
|
|
|
|
Fig. 4 : Orecchie a trombetta ed encondromi. |
Un bambino di 13-mesi con sintomi di BOR. L’orecchio sinistro appositamente sviluppato; all’interno l’orecchio destro, il canale uditivo e atresia lobo impropriamente sviluppati. |
|
|
|
|
Fig. 5 : Micrococlea in una sindrome branchio-oto-renale (BOR). |
|
|
|
|
|
Fig. 6 : Residuo branchiale cervicale |
Ragazza con la sindrome di BOR, fistola carotidea bilaterale |





Diagnosi
Una storia familiare positiva per brachiale, otologica, e le malformazioni renali è suggestivo di sindrome di BOR.
Se nessuna storia familiare di malattia è noto, criteri clinici possono essere utilizzate per fare la diagnosi per pazienti con almeno tre criteri principali. In alternativa, la diagnosi può essere fatta per i pazienti che dimostrano due criteri principali e due criteri minori.
Criteri principali
1. Malformazione dei padiglioni auricolari
2. Perdita dell’udito
3. Encondromi preauricolari
4. Anomalie renali
5. Anomalie secondarie dell’arco branchiale
6. Stenosi del canale uditivo esterno
Criteri minori
1. Appendici preauricolari
2. Aplasia del dotto lacrimale
3. Anomalie dell’orecchio medio
4. Anomalie dell’orecchio interno
5. Malformazioni del palato o asimmetria facciale
6. Gozzo eutiroideo
Giudizio fenotipico
Otologico
Le manifestazioni otologiche della BOR sono i sintomi più comuni, con oltre il 90% delle persone in almeno uno dei seguenti:
1. La sordità può presentarsi come trasmissiva, neurosensoriale o mista. La gravità della sordità è variabile, che vanno dalla perdita dell’udito da lieve a profonda. Inoltre, l’entità della perdita dell’udito può essere stabile o progressiva. La perdita dell’udito è ormai consolidata come il tratto singolo più comune tra quelli con la sindrome BOR, stimata in 89% degli individui in uno studio di Fraser et al. Tra quelli con perdita dell’udito, il 50% presentava ipoacusia mista, 30% trasmissiva de il 20% neurosensoriale. E ‘stato inoltre stimato che il 2% dei bambini con sordità profonda hanno BOR.
2. Deformità del padiglione .
3. Bozzi preauricolari.
4. Appendici preauricolari.
5 Stenosi del condotto uditivo esterno:
6. Malformazioni degli ossicini dell’orecchio medio, quali ipoplasia o spostamento
7 Orecchio interno: coclea ipoplasica, canali semicircolari laterali displasici, l’allargamento degli acquedotti vestibolari o cocleari.
Anomalie Arco Branchiale
1. Cisti branchiali leporine.
2. Brancial tratto del seno leporino.
3. Branchiale fistole leporino.
Malformazioni renali
1. Uno spettro di malformazioni renali è possibile, da ipoplasia al agenesia.
2.Cisti caliceale.
3. Uretero-pelvica ostruzione del giunto.
4. Reflusso vescico-ureterale
Altri risultati associati
1. Prolasso della valvola mitrale
2. Palatoschisi
3. Aplasia del dotto lacrimale
4. Paralisi del nervo facciale
5. Retrognazia
Studi dei Geni
Attualmente, vi sono tre mutazioni genetiche conosciuti che provocano sindrome BOR. Se viene effettuata una diagnosi clinica di sindrome di BOR, la conferma dovrebbe essere tentato attraverso l’analisi di sequenza per EYA1. Se una mutazione EYA1 non viene trovato con l’analisi di sequenza, si dovrebbe utilizzare l’analisi duplicazione / cancellazione per EYA1 e analisi di sequenza di SIX 1 e SIX 5.
Mutazione EYA1
· Circa il 40% delle persone con sindrome di BOR avrà una mutazione EYA1.
· EYA1 codifica per un cofattore trascrizione che si esprime nel mesenchima metanefrico durante lo sviluppo dei reni.
SIX1 mutazione
· La mutazione SIX1 è stimata essere trovato 2% dei casi di sindrome BOR.
· Codifica fattori di trascrizione che controllano l’espressione di GDNF e PAX2.
· La maggior parte dei pazienti con questa mutazione non dimostrano malformazione renale.
SIX5 mutazione
· La mutazione SIX5 è presente in circa il 2,5% dei casi di sindrome di BOR.
· Udienza può essere normale in questi pazienti.
Gestione
Valutazione Otologic
· Le prove devono comprendere ABR, audiometria tonale, e la prova di emissione.
· Imaging delle ossa temporali con una TAC è giustificata.
· Una valutazione annuale uditivo dovrebbe essere suggerito.
Valutazione dell’Arco Branchiale
· L’esame fisico seguita dalla tomografia computerizzata (TC), fistologramma, o risonanza magnetica (MRI) se le masse sono palpate .
Valutazione renale
· Ecografia renale
· BUN e creatinina
· I pazienti devono seguire un follow-up annualmente con la nefrologia e urologia.
La consulenza genetica
· È importante che i pazienti affetti da BOR tengano conto conto che c’è una probabilità del 50% di trasmissione per ogni bambino a causa autosomica dominante.
· Test prenatale per BOR è disponibile per quelli con una storia familiare positiva.
Trattamento
Anomalie otologiche
· Rilevare eventuale ipoacusia, utilizzare il trattamento acustico appropriato come indicato dalla gravità della perdita, come la protesi o l’impianto cocleare dell’udito.
· Se l’orecchio medio è intatto, con stenosi del canale uditivo esterno, si può prendere in considerazione una canaloplastica.
· Considerate le procedure di chirurgia plastica per la deformità della Pinna.
Anomalie branchiali
· Asportazione di cisti, fistole, o del seno, con dissezione per tutta la lunghezza del tratto e la tonsillectomia.
Anomalie renali
· Il trattamento dipende dalla gravità delle complicanze renali, ma sia la gestione chirurgica e medica dovrebbe essere utilizzato.
· Il trapianto renale può essere considerato se la malattia renale allo stadio terminale si sviluppa.
Manifestazioni generali
La displasia renale è riportato in oltre due terzi dei pazienti e varia da pali rastremati superiori (duplicazione del sistema di raccolta) alla marcata agenesia del rene. La sindrome può includere sequenza Potter.
Dove posso trovare ulteriori informazioni sulla sindrome branchiootorenale?
Si possono trovare le seguenti risorse sulla sindrome branchiootorenal utili. Questi materiali sono scritti per il pubblico in generale.
• MedlinePlus – Informazioni sanitarie (4 collegamenti)
• Malattie Genetiche e Rare Information Center – Informazioni sulle malattie genetiche e le malattie rare
• Ulteriori NIH Risorse – National Institutes of Health
National Institute on Deafness e altri disturbi della comunicazione (NIDCD)
• Risorse educative – Pagine di informazioni (5 collegamenti)
• Supporto paziente – Per i pazienti e le famiglie (3 collegamenti)
Potreste essere interessati a queste risorse, che sono progettati per gli operatori sanitari e ricercatori.
• Gene Recensioni – Sintesi clinica
• Test genetici Registry – archivio di informazioni test genetico (2 collegamenti)
• PubMed – La letteratura recente
• OMIM – Catalogo Malattia genetica (2 collegamenti)
Quali altri nomi la gente usa per la sindrome branchiootorenale?
• BOR
• Sindrome BOR
• Displasia Branchiootorenale
• Branchio-Otorenale Displasia
• Branchio-Otorenale Syndrome
• Sindrome Branchio-Oto-Renale
• Sindrome di Melnick-Fraser
Per ulteriori informazioni sulla denominazione condizioni genetiche, vedere la Genetics Home Reference Condizione Linee guida di denominazione e di come sono condizioni genetiche e geni denominati? nel manuale.
Cosa succede se ho ancora domande specifiche sulla sindrome branchiootorenal?
Chiedi alla Genetica e Malattie Rare Information Center .
References/Suggested Reading
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, Weil D, Cruaud C, Sahly I, Leibovici M,
Amer I, Falzon A, Choudhury N, Ghufoor K. Branchiootic syndrome-a clinic case report and review of the literature. J Pediatr Surg . 2012;47:1604-1606.
Bitner-Glindzicz M, Francis M, Lacombe D, Vigneron J, Charachon R, Boven K, Bedbeder P, Van Regemorter N, Weissenbach J, Petit C. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat Genet1997;15:157–64.[CrossRef][Medline][Web of Science]
Blanck C, Kohlhase J, Engels S, Burfeind P, Engel W, Bottani A, Patel MS, Kroes HY, Cobben JM. Three novel SALL1 mutations extend the mutational spectrum in Townes-Brocks syndrome: clinical and genetic implications. Am J Hum Genet2000;66:1715–20.[CrossRef][Medline][Web of Science]
Borsani G, DeGrandi A, Ballabio A, Bulfone A, Bernard L, Banfi S, Gattuso C, Mariani M, Dixon M, Donnai D, Metcalfe K, Winter R, Robertson M, Axton R, Brown A, Van Heyningen V, Hanson I. EYA4, a novel vertebrate gene related to Drosophila eyes absent. Hum Mol Genet1999;8:11–23.[Abstract/FREE Full text]
Buck A, Kispert A, Kohlhase J. Embryonic expression of the murine homologue of SALL1, the gene mutated in Townes-Brocks syndrome. Mech Dev2001;104:143–6.[CrossRef][Medline][Web of Science]
Chen A, Francis M, Ni L, Cremers C, Kimberling W, Sato Y, Phelps P, Bellman S, Wagner M, Pembrey M, Smith RJ. Phenotypic manifestations of branchiootorenal syndrome. Am J Med Genet . 1995;58:365-370.
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoot VS, Schwartz CE, Smith RJ. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat . 2004;23:582-9. PubMed citation![]()
Correa-Cerro LS, Kennerknecht I, Just W, Vogel W, Müller D. The gene for branchio-oculo-facial syndrome does not colocalize to the EYA1-4 genes. J Med Genet2000;37:620–3.[FREE Full text]
Engels S, Kohlhase J, McGaughran J. A SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype. J Med Genet2000;37:458–60.[FREE Full text]
Fraser FC, Sproule JR, Halal F. Frequency of the branchio-oto-renal (BOR) syndrome in children with profound hearing loss. Am J Med Genet . 1980;7:341-349.
Fujimoto A, Lipson M, Lacro RV, Shinno NW, Boelter WD, Jones KL, Wilson MG. New autosomal dominant branchio-oculo-facial syndrome. Am J Med Genet1987;27:943–51.[CrossRef][Medline][Web of Science]
Gene Review: Branchiootorenal Spectrum Disorders This link leads to a site outside Genetics Home Reference. Kochhar A, Fischer SM, Kimberling WJ, et al. Branchio-oto-renal syndrome. Am J Med Genet. 2007;15(143A(14)):1671-8, Review. PubMed citation![]()
Kumar S, Kimberling WJ, Weston MD, Schaefer BG, Berg MA, Marres HA, Cremers CW. Identification of three novel mutations in human EYA1 protein associated with branchio-oto-renal syndrome. Hum Mutat1998;11:443–9.[CrossRef][Medline][Web of Science]
Kumar S, Deffenbacher K, Marres HAM, Cremers CWRJ, Kimberling WJ. Genomewide search and genetic localization of a second gene associated with autosomal dominant branchio-oto-renal J Hum Genet2000;67(suppl 2):401.
Legius E, Fryns JP, Van den Berghe H. Dominant branchial cleft syndrome with characteristics of both branchio-oto-renal and branchio-oculo-facial syndrome. Clin Genet1990;37:347–50.[Medline]
Kumar S, Marres HAM, Cremers CWRJ, Kimberling WJ. Mutation analysis of EYA1 gene associated with
branchio-oto-renal syndrome and refining the BOR2 region on chromosome 1q. Am syndrome. J Med Genet2000;37:303–7.[FREE Full text]
Lin AE, Doherty R, Lea D. Branchio-oculo-facial and branchio-oto-renal syndromes are distinct entities. Clin Genet1992;41:221–3.[Medline]
Lin AE, Gorlin RJ, Lurie IW, Brunner HG, Van der Burgt I, Naumchik IV, Rumyantseva NV, Stengel Rutkowski S, Rosenbaum K, Meinecke P, Müller D. Further delineation of the branchio-oculo-facial syndrome. Am J Med Genet1995;56:42–59.[CrossRef][Medline][Web of Science]
Lin AE, Semina EV, Daack-Hirsch S, Roeder ER, Curry CJ, Rosenbaum K, Weaver DD, Murray JC. Exclusion of the branchio-oto-renal syndrome locus (EYA1) from patients with branchio-oculo-facial syndrome. Am J Med Genet2000;91:387–90.[CrossRef][Medline]
McKusick VA. Mendelian inheritance in man. Catalogs of human genes and genetic disorders. 12th ed. Baltimore: Johns Hopkins University Press, 1998.
Mishima N, Tomarev S. Chicken eyes absent 2 gene: isolation and expression pattern during development. Int J Dev Biol1998;42:1109–15.[Medline][Web of Science]
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, Kumar S, Neuhaus TJ, Kemper MJ, Raymond RM Jr, Brophy PD, Berkman J, Gattas M, Hyland V, Ruf EM, Schwartz C, Chang EH, Smith RJ, Stratakis CA, Weil D, Petit C, Hildebrandt F. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes.Proc Natl Acad Sci US A. 2004 May 25;101(21):8090-5.Epub 2004 May 12. PubMed citation![]()
Sahly I, Andermann P, Petit C. The zebrafish eya1 gene and its expression pattern during embryogenesis. Dev Genes Evol1999;209:399–410.[CrossRef][Medline][Web of Science]
Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 1989. Sanggaard KM, Rendtorff ND, Kjaer KW, Eiberg H, Johnsen T, Gimsing S, Dyrmose J, Nielsen KO, Lage K, Tranebjaerg L. Branchio-oto-renal syndrome: detection of EYA1 and SIX1 mutations in five out of six Danish families by combining linkage, MLPA and sequencing analyses.Eur J Hum Genet.2007 Nov;15(11):1121-31.Epub 2007 Jul 18. PubMed citation![]()
Sobel E, Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet1996;58:1323–37.[Medline][Web of Science]
Vincent C, Kalatzis V, Abdelhak S, Chaib H, Compain S, Helias J, Vaneecloo FM, Petit C. BOR and BO syndromes are allelic defects of EYA1. Eur J Hum Genet1997;5:242–6.[Medline][Web of Science]
Xu PX, Cheng J, Epstein JA, Maas RL. Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci USA1997;94:11974–79.[Abstract/FREE Full text]
Wang Y, Treat K, Schroer RJ O’Brien JE, Stevenson RE, Schwartz CE (giugno 1994). “Localizzazione di (BOR) sindrome Branchio-oto-renale a una regione del cromosoma 3 Mb 8q”. Am. J. Med.. Genet. 51 (2):. 169-75
Sindrome di Stickler/ sindrome di Marshall ,artro-oftalmopatia progressiva ereditaria , displasia di Stickler
distrofia ereditaria artro-oftalmologica
– trasmissione autosomica dominante
– espressività variabile
– sordità neurosensoriale-mista
– palatoschisi
– insufficienza muscolatura faringo-laringea
– miopia
|
Sindrome di Stickler
|
Modalità di trasmissione |
Tipo di deficit uditivo |
Anomalie associate |
|
|
A.D. Gene COL2A1(cr. 12) tipo I, COL11A1 (cr. 1) tipo II, COL11A2 (A.D. e A. R.) |
Ipoacusia neurosensoriale bilaterale piú marcata sulle frequenze acute |
Anomalie ossificazione (epifisi,diafisi), anomalie articolari, ipoplasia facciale, grave miopia (insorgenza primo anno di vita) e frequente distacco di retina, palatoschisi (occasionale). |
CHE COSA È LA SINDROME DI STICKLER??PAG.1
TIPI, PAG.1
STORIA, PERCHÉ SINDROME DI STICKLER?, PAG.2
CAUSE GENETICHE PAG.3
QUALI GENI SONO LEGATI ALLA SINDROME DI STICKLER? PAG.3
EPIDEMIOLOGIA, PAG.4
CAUSE, PAG.4
SINTOMI, PAG.4
LA DIAGNOSI DIFFERENZIALE,PAG10
APPROFONDIMENTI, PAG 17
CHE COSA È LA SINDROME DI STICKLER?
La Sindrome di Stickler (artro-oftalmopatia ereditaria progressiva) è un gruppo di malattie genetiche che interessano il tessuto connettivo , in particolare il collagene . La sindrome di Stickler è un sottotipo di collagenopatia, tipo II e XI Due geni sono stati individuati come responsabili di questa sindrome (COL2A1, COL11 A2), entrambi implicati nella costituzione di certi tipi di collagene. La sindrome può rivelarsi alla nascita con una palatoschisi. micrognatismo, ptosi linguale e soprattutto incompetenza della muscolatura faringo-laringea (turbe della deglutizione ed ostruzione respiratoria) Nelle due varianti cliniche della sindrome, (I e III) è anche presente una forte miopia Durante l’accrescimento diventano evidenti anomalie scheletriche e cartilaginee a carico degli arti. La sordità è neurosensoriale o mista, talvolta complicata da esiti di fiogosi timpaniche croniche.. La sindrome Stickler è caratterizzata da peculiari anomalie facciali, problemi oculari, perdita dell’udito e problemi articolari. E ‘stata studiato per prima e caratterizzato da Gunnar B. Stickler nel 1965.
TIPI
Cambiamenti genetici sono legate ai seguenti tipi di sindrome di Stickler (Annunen S, ed Al 1999; Liberfarb RM ed Al,2003)
Sindrome di Stickler, COL2A1 (75% dei casi Stickler) Fig.1
Sindrome di Stickler, COL11A1
Sindrome di Stickler, COL11A2 (non-oculare)
Sindrome di Stickler, COL9A1 (variante recessiva)
Se ci sono due o tre tipi di sindrome di Stickler è controversa. Ogni tipo è qui presentati per il gene coinvolto. La classificazione di queste condizioni sta cambiando i ricercatori a saperne di più circa le cause genetiche.
|
|
Fig.1 sindrome di Stickler tipo 1 |

I ricercatori hanno descritto diversi tipi di sindrome di Stickler, che si distinguono per la loro causa genetica e loro segni e sintomi caratteristici. In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma IL tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Tipi II e III sono più probabili di tipo I per aver perdita dell’udito. I tipi IV e V sono molto rari e sono stati diagnosticati solo in alcuni individui.
Una condizione simile chiamata sindrome di Marshall è caratterizzata da un aspetto caratteristico del viso, anomalie oculari, perdita dell’udito e artrosi precoce. La Sindrome di Marshall può includere anche bassa statura, che tipicamente non si riscontra nei pazienti con sindrome di Stickler. Se la sindrome di Marshall rappresenta una variante della sindrome di Stickler o una patologia separata è controversa.
Perché sindrome di Stickler?
Stickler sindrome prende il nome dal dottor Gunnar B Stickler. Nel 1960 un ragazzo di dodici anni è stato esaminato presso la Fondazione Mayo nel Minnesota, USA. Il ragazzo aveva ingrandimenti ossee diverse articolazioni ed è stato estremamente miope. Sua madre era totalmente cieco. Dr Stickler ha scoperto che c’erano altri membri della famiglia con sintomi simili, il primo membro della famiglia essendo stati visti dal dottor Charles Mayo nel 1887. Questo ha spinto il dottor Stickler per studiare la famiglia. Con colleghi ha lavorato per definire la condizione, i risultati in corso di pubblicazione nel giugno 1965. Dr Stickler provvisoriamente chiamato la condizione ereditaria PROGRESSIVE ARTRO-OFTALMOPATIA conosciuta in tutto il mondo, come la sindrome di Stickler.
CAUSE GENETICHE
La sindrome deriva da una mutazione di diversi geni del collagene durante lo sviluppo fetale. Si tratta di un sindrome , indipendente dal sesso, autosomica dominante Fig.2, il che significa che un paziente con la sindrome ha una probabilità del 50% di passare la malattia ad ogni figlio. Ci sono tre varianti di Stickler sindrome identificati, ciascuno associato ad un gene di biosintesi del collagene. Un difetto metabolico riguardante l’acido ialuronico e il collagene di tipo 2-d si presuma sia la causa di questa sindrome.
|
|
Fig.2 La sindrome di Stickler è ereditata come modello autosomico dominante |

QUALI GENI SONO LEGATI ALLA SINDROME DI STICKLER?
Le mutazioni nei geni COL11A1, COL11A2 e COL2A1 COL9A1, COL9A2 e geni causano tipi di sindrome di Stickler da I a V, rispettivamente. La sindrome di Marshall, che può essere una variante di Stickler sindrome, risulta da mutazioni nel gene COL11A1. Perché ci siano due o tre tipi di sindrome è controverso. Ciascun tipo è qui presentato secondo il gene in causa. La classificazione di tali condizioni cambierà quando i ricercatori apprenderanno di più circa le cause genetiche. Tali geni sono coinvolti nella produzione del collagene tipo II tipo IX e tipo XI. I collageni sono molecole complesse che provvedono alla struttura e alla resistenza del tessuto connettivo che sostengono le articolazioni del corpo e degli organi. Il collagene di tipo II, tipo IX e XI tipo sono componenti del vitreo, cartilagine e altri tessuti connettivi.. Una mutazione in uno di questi geni interrompe la produzione, l’elaborazione o la formazione del collagene tipo II o XI. Le molecole difettose di collagene o la ridotta quantità di collagene influisce sullo sviluppo di ossa e altri tessuti connettivali, portando al caratteristico quadro della Stickler.
Altri geni, sebbene sconosciuti, possono causare la sindrome perché non tutti i soggetti colpiti hanno mutazioni in uno dei tre geni già identificati. La sindrome di Stickler tipo 1 è dovuta alle mutazioni del gene COL2A1 (12q13.11-q13.2), quella tipo 2 alle mutazioni dei geni COL11A1 (1p21) e COL11A2 (6p21.3). Inoltre, in una famiglia del Marocco, è stata osservata una forma autosomica recessiva associata alle mutazioni del gene COL9A1 (6q12-q14). Le mutazioni che si verificano nel primo gruppo di geni sono a carattere trasmissivo autosomico dominante mentre quelle a carico del secondo gruppo sono a carattere autosomico recessivo e, in entrambi i casi, le conseguenze fenotipiche che comportano sono molte.
EPIDEMIOLOGIA
Complessivamente la prevalenza stimata di sindrome Stickler è circa 1 su 10.000. Colpisce 1 neonato su 7.500-9.000. Alcuni medici credono che ben 3 in 10.000 persone sono colpite, ma ulteriori ricerche sono necessarie per confermare questo. Come una malattia ereditaria, la sindrome di Stickler è normalmente passata di padre in figlio. C’è una probabilità del 50% dei bambini essere colpiti in questo modo anche se ci sono alcuni casi registrati dove si è verificato per la prima volta in un bambino.
CAUSE
La sindrome è ereditaria con schema autosomico dominante. Si suppone che insorga da una mutazione di diversi geni del collagene durante lo sviluppo fetale. È indipendente dal sesso, cioè i soggetti affetti dalla sindrome hanno la probabilità del 50% di passarla a ciascun figlio.
SINTOMI
Gli individui con sindrome di Stickler hanno una serie di segni e sintomi. Alcune non hanno segni e sintomi; altri hanno alcune o tutte le funzioni descritte di seguito. Inoltre, ogni caratteristica di questa sindrome può variare da una forma lieve ad una grave.
Fu studiata e catalogata per la prima volta dal dr. G.B. Stickler nel 1965. È caratterizzata dal
1) tipico aspetto facciale appiattito, Fig. 3
2) patologie oculari,
3) perdita dell’udito,
4)disturbi alle articolazioni.
1)tipico aspetto facciale appiattito: Fig. 3A-B-C-D-E questo è causato da ossa sottosviluppate del centro del viso, compresi gli zigomi e il ponte del naso. Un gruppo particolare di caratteristiche fisiche, chiamata sequenza di Pierre Robin, è comune anche in persone con sindrome di Stickler. La sequenza Robin include un’apertura nel tetto della bocca (palatoschisi), una lingua piccola , che è posto più indietro rispetto al normale (glossoptosi), e una piccola mandibola (micrognazia). Questa combinazione di caratteristiche può portare a problemi di alimentazione e difficoltà di respirazione.. Si può rivelare alla nascita tramite una schisi velopalatina, completa o sottomucosa, integrantesi talvolta in una sequenza di Pierre Robin: triade schisi palatina/microretrognazia/glossoptosi e, soprattutto, incompetenza dell’incrocio faringolaringeo, fonte di disturbi della deglutizione e di ostruzione respiratoria che possono richiedere una tracheotomia transitoria (la sindrome di Stickler è una causa ormai ben nota della sequenza di Pierre Robin). Il dismorfismo faciale è costante (ipoplasia del piano medio del volto), ma spesso difficile da valutare nel lattante.
|
|
|
||||
|
Fig. 3A-B-C-D-E |
|
||||
|
|
||||
|
|




2)patologie oculari,
Miopia
Alto rischio di distacchi di retina, che possono influenzare entrambi gli occhi.
Cataratta
Glaucoma
La sindrome di Stickler è una vitreo-retinopatia. Anomalie di formazione vitreo e dell’architettura gel sono patognomonico della sindrome di Stickler e, a nostro avviso un prerequisito per la diagnosi. 6 Due distinti fenotipi possono essere riconosciute. 7-9La maggior parte dei pazienti hanno una caratteristica anomalia congenita del vitreo (il tipo 1 fenotipo, Fig 4 A ) e questo correla con difetti di di procollagene di tipo II.78 Un gel vitreale apparentemente rudimentale occupa lo spazio immediato retrolentale ed è delimitato da una membrana distinta piegata. In una minoranza di pedigree c’è un fenotipo diverso con fasci radi e irregolarmente ispessite di fibre in tutta la cavità vitrea (tipo 2 fenotipo, Fig. 4B).
|
|
|
|
Fig4 A : fenotipo tipo 1, |
Fig. 4B :fenotipo, tipo 2 |


In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma il tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Molte persone con sindrome di Stickler sono molto miopi (descritto ed aventi elevata miopia ) a causa della forma dell’occhio. Anomalie dello sviluppo dell’angolo di drenaggio camera anteriore predispongono i pazienti a glaucoma, 10Fig. 7 (b), ma la complicanza più grave oftalmica riferisce l’elevato rischio di distacco di retina, di solito come conseguenza di gigante formazione di rottura retinica Può Anche presente come complicazione la cataratta associata alla sindrome di Stickler. La sostanza gelatinosa all’interno dell’occhio (il vitreo ) ha un aspetto distintivo dei tipi di sindrome di Stickler associati ai geni COL2A1 e COL11A1. Come risultato si consiglia appuntamenti fissi ad un oculista specialista. Il tipo di sindrome di Stickler associata al gene COL11A2 non influenza l’occhio. [2]
|
|
|
Tutti i pazienti hanno mostrato il fenotipo vitreo “perline” (Fig. 4 C-D-E) ed hanno confermato le mutazioni nella catena α1 di tipo XI collagene ( COL11A1 ) |
|
|
|
Fig. 5 A-B-C Caratteristica cataratta corticale |
|
|
|
Fig. 6 (A) lieve opacità della capsula posteriore del cristallino dell’occhio destro osservata inizialmente all’età di 7 anni. (B) Aspetto del fondo dimostrare la retina alterazioni della pigmentazione della periferia e alterazioni degenerative della retina all’interno del polo posteriore. (C) opacizzazione marcata e la fusione delle capsule lente nell’occhio di destra osservato a 9 anni di età. (D) Lievi opacità della capsula si osservano ancora due anni più tardi, a 11 anni di età. L’errore di rifrazione in questa fase è +1.25 e l’acuità visiva per una distanza di 6/12 (20/40). |



3)perdita dell’udito. La sordità di percezione, trasmissione o mista è incostante, e spesso mascherata o aggravata dai problemi di otite cronica che sono associati all’incompetenza faringea. Essa è spesso evolutiva. Sono descritti tre tipi di sindrome di Stickler: nei tipi 1 e 3, agli altri elementi della sindrome è associata una miopia molto forte con rischio di degenerazione vitro-retinica. L’esame oftalmologico di ogni bambino sordo e di ogni lattante con incompetenza faringolaringea deve permettere d’individuare questa sindrome e correggere precocemente la miopia. La maggior parte delle persone con sindrome di Stickler hanno anomalie scheletriche che colpiscono le articolazioni. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
4)disturbi alle Ossa e Articolazioni
Caratterizzata dall’associazione tra segni oculari, correlati alle forme più o meno complete della sequenza di Pierre Robin (si veda questo termine), alterazioni scheletriche e sordità neurosensoriale (10% dei casi) .Questi segni e sintomi variano ampiamente tra gli individui affetti. Questa sindrome è dovuta a una alterazione delle catene ![]() di alcuni collageni. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare di tipo artrosico o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
di alcuni collageni. Sono anche presenti delle anomalie scheletriche e cartilaginee, che possono portare a una piccola statura o, al contrario, a una grande statura. Le articolazioni dei bambini affetti e giovani adulti possono essere allentati e molto flessibile (ipermobile), anche se le articolazioni diventano meno flessibile con l’età. Artrite appare spesso presto nella vita e può causare dolore articolare di tipo artrosico o rigidità. Possono verificarsi anche problemi con le ossa della colonna vertebrale (vertebre), tra cui curvatura anomala della colonna vertebrale (scoliosi o cifosi) e appiattito vertebre (platispondilia). Queste anomalie della colonna vertebrale possono causare mal di schiena.
I ricercatori hanno descritto diversi tipi di sindrome di Stickler, che si distinguono per la loro causa genetica e loro segni e sintomi caratteristici. In particolare, le anomalie oculari e gravità della perdita uditiva differiscono tra i tipi. Il tipo I ha il più alto rischio di distacco di retina. Il tipo II include anche anomalie oculari, ma il tipo III non ha interessamenti oculari (ed è spesso chiamato sindrome di Stickler non-oculare). Nei tipi II e III la perdita dell’udito è più probabili rispetto al tipo I . I tipi di IV e V sono molto rari e sono stati diagnosticati solo in alcuni individui.
Una simile condizione chiamata sindrome di Marshall è caratterizzata da un aspetto caratteristico del viso, anomalie oculari, perdita dell’udito e artrosi precoce. Sindrome di Marshall può includere anche bassa statura, che tipicamente non si riscontra nei pazienti con sindrome di Stickler. E’ controverso se la sindrome di Marshall rappresenti una variante della sindrome di Stickler o piuttosto n’entità a se.
|
|
|
|
|
Fig. 7 (A) Miopia. |
Fig. 7 (b) Foto che mostra vasi congiuntivali dilatati a bordo corneale (flush ciliare, filo circumcorneale) e cornea nebbiosa caratteristica di glaucoma acuto ad angolo chiuso |
|
|
|
|
|



Le persone con sindrome di Stickler accusano una serie di segni e sintomi, dalla paucità alla grave espressione fenotipica.
Una tipica caratteristica della sindrome è l’aspetto della faccia, come appiattito. Ciò è causato dalle ossa poco sviluppate nel centro della faccia, comprese le mascelle e il setto del naso.
Molti pazienti con la sindrome di Stickler soffrono di la cataratta giovanile, miopia grave Fig. 7 (A) , a causa della forma dell’occhio. Gli individui con gli occhi colpiti dalla patologia sono soggetti a pressione oculare aumentata (glaucoma) e a degenerazione vitreo-retinica o corio-retinica, a distacco della retina e l’uveite cronica.. La sostanza gelatinosa dentro l’occhio ha un aspetto caratteristico nella sindrome di tipo COL2A1 e COL11A1. Quella di tipo COL11A2 non colpisce l’occhio.
I soggetti con questa patologia hanno disturbi che colpiscono anche altri organi. Artrite, anormalità dell’estremità delle ossa lunghe, anomalia delle vertebre, curvatura della spina dorsale, gibbosità, dolore all’articolazioni,
ginocchio valgo sono tutti problemi che possono occorrere alle ossa e alle articolazioni. L’artrite spesso compare a giovane età e peggiora con la crescita).Le articolazioni dei bambini colpiti e dei giovani possono essere molto flessibili (ipermobili).. Difficoltà nell’apprendimento possono anche avvenire per la menomazione all’udito e alla vista.Tipico di soggetti con Stickler può includere guance piatte, setto nasale appiattito e mascella piccola, anomalie del palato.
Un altro segno della sindrome è la diminuzione dell’udito da lieve a grave che, per alcuni, può essere progressiva. La perdita di udito è stata riscontrata nel 62,9 per cento dei casi, in una forma lieve o moderata. Il deficit uditivo era principalmente neurosensoriale ovvero a carico della coclea o del nervo acustico (67,8 per cento).
Il deficit trasmissivo, danno a carico dell’orecchio esterno o medio, è risultato essere meno frequente (14,1 pe cento) cosi come quello misto (18,1 per cento). Le forme di ipoacusia trasmissiva e mista sono state riscontrate principalmente nei giovani pazienti o nei pazienti con un difetto al palato.
Complessivamente le mutazioni a carico di COL11A1 (82,5 per cento) e COL11A2 ( 94,1 per cento) risultano più frequentemente associate con la menomazione uditiva, rispetto alle mutazioni di COL2A1 ( 52,2 per cento Considerando questi dati si può sostenere che la menomazione uditiva nei pazienti affetti da sindrome di Stickler è comune e che, nonostante la forma di ipoacusia che predomini sia quella neurosensoriale, anche l’ipoacusia trasmissiva e mista possono verificarsi.
Le mutazioni dei diversi geni codificanti per il collagene sono associate a diversi tassi di prevalenza di ipoacusia ma esistono ancora un gran numero di variazioni fenotipiche che non sono state associate a specifiche caratteristiche genotipiche.
Vista la portata del problema il consiglio è di eseguire un regolare follow-up dell’udito nei pazienti affetti da sindrome di Stickler, valutazioni audiologiche ogni sei mesi fino all’età cinque anni, poi annualmente,
La diagnosi differenziale
Diversi disturbi simili alla sindrome di Stickler sono stati descritti e il loro status di entità distinte rimane controversa. Dati genetici molecolari stanno cominciando a informare questo dibattito ma resta incertezza. Questo sarà risolto solo quando più dati di genotipo diventano disponibili in combinazione con la descrizione dettagliata dei oculare associata e fenotipo non-oculare.
SINDROME DI WAGNER
Wagner29ha registrato una grande famiglia svizzera con un disordine autosomico dominante dell’occhio assomiglia sindrome di Stickler, ma senza il distacco di retina. Molte delle famiglie successivamente segnalato come sindrome di Wagner hanno avuto caratteristiche sistemiche in comune con la sindrome di Stickler e la distinzione tra le due condizioni è diventata sfocata. Infatti, alcuni autori hanno suggerito che essi sono la stessa malattia. 30La prova che le famiglie che mostrano solo le manifestazioni oculari della sindrome di Wagner hanno una condizione distinta dalla sindrome di Stickler è venuto dalla constatazione di linkage to 5q13-q14 nella famiglia di origine Wagner 31e esclusione di linkage to COL2A1 in un’altra famiglia.32Alla luce di questi risultati, il termine “sindrome di Wagner-Stickler” dovrebbe essere abbandonato. Korkko et al33ha riferito di un paziente con la sindrome di Wagner derivante da una sostituzione di ingombranti aspartato aminoacido glicina per nell’esone 10 di COL2A1 e postulato un possibile legame tra il tipo di mutazione e fenotipi Stickler o Wagner. Tuttavia, frequente distacco della retina e, in misura minore cataratta sono stati attribuiti alla sindrome di Wagner, mentre nel report originale 29nessun paziente ha subito un distacco della retina, “cataracta Complicata” è stata quasi universale, e la miopia era bassa in tutti i casi. Si potrebbe sostenere che nella famiglia riportata da Korkko et al 33fenotipo assomiglia più da vicino la sindrome di Stickler sindrome di Wagner.
EROSIVA VITREORETINICA
Brown et al 34 hanno descritto un disordine autosomico occhio dominante hanno chiamato vitreoretinica erosiva con un fenotipo simile sindrome di Wagner e privo di eventuali anomalie sistemiche. Questa condizione è stato mappato a 5q13-q14 suggerendo che potrebbe essere una variante allelica della sindrome di Wagner. 31
SINDROME DI MARSHALL
Marshall 35ha registrato una grande famiglia che mostra ereditarietà autosomica dominante della cataratta, miopia, vitreo anormale, ipoplasia mediofacciale e sordità congenita. Marshall ha pensato il fenotipo potrebbe rappresentare una forma incompleta di ereditaria anidrotica displasia ectodermica, ma ha riconosciuto che il pelo era normale e la prova di ipodonzia e ipoidrosi “non era fortemente convincente”. Shanske et al 36ha notato dalle foto pubblicate che uno dei pazienti di Marshall era colpito da ipertelorismo con ipertelorismo forse lieve in altri. C’è stato molto dibattito se la sindrome di Marshall è una entità distinta 37 e, in caso affermativo, se displasia ectodermica è una caratteristica della condizione. 36Ayme e Preus 38 effettuate cluster analysis sui rapporti pubblicati di Marshall e Stickler sindrome pazienti e ha concluso che erano diversi. Resta da vedere se questo problema può essere risolto con l’analisi genetica molecolare. Griffith et al 39hanno riportato una mutazione COL11A1 in una famiglia ha detto di avere la sindrome di Marshall. Shanske et al40hanno suggerito la famiglia aveva la sindrome di Stickler, ma gli autori hanno risposto che, come riportato da Marshall famiglia di origine, i loro pazienti avevano cataratta congenita e giovanile, vitreo fluido, perdita di udito, e simili aspetto cranio-facciale e la radiologia. 41,in quanto questi sono tutte le caratteristiche della sindrome di Stickler riconosciuto e non vi è alcuna informazione sul fenotipo vitreo, il problema rimane irrisolto.
SINDROME DI WEISSENBACHER-ZWEYMÜLLER E OSMED
Weissenbacher e Zweymuller 42hanno descritto un neonato con la sequenza di Pierre-Robin, naso camuso, prossimale accorciamento dell’arto, femori a forma di campana muta e omeri, e schisi vertebrali coronali. I genitori erano sani e non collegati. Giedion et al 43hanno seguito lo stesso paziente a 18 anni di età. Sordità neurosensoriale aveva sviluppato all’età di 5. By adulta accorciamento della vita arto era del tutto risolto e l’altezza e l’aspetto erano sostanzialmente normale. Non c’era alcuna anomalia occhio. Epifisi ingrossati erano una caratteristica radiologica di primo piano in adolescenza. Giedion et al 43hanno riferito di altri tre pazienti con lo stesso fenotipo e ha coniato il nome OtoSpondiloMegaEpifiseale Displasia (OSMED). Essi hanno concluso che la sindrome Weissenbacher-Zweymuller (WZS) e OSMED erano la stessa sindrome. Pihlajamaa et al 44,successivamente è emerso che il paziente WZS originale era eterozigoti per una mutazione in COL11A2. Sono state descritte altre famiglie mostrano autosomica dominante di un simile non-oculare Stickler sindrome fenotipo come risultato di mutazioni COL11A2. 45-47 Van Steensel et al 48riportano tre fratelli dei genitori consanguinei che erano omozigoti per una mutazione COL11A2 e aveva il OSMED fenotipo. Vikkula et al 46hanno riportato diversi membri di una famiglia consanguinea che erano omozigoti per una mutazione in COL11A2. Avevano una grave sordità neurosensoriale congenita, ipoplasia medio-facciale, a breve, naso all’insù, gli occhi prominenti, creste sopraorbitali prominenti, e insorgenza precoce adulta di grave artrosi delle anche, ginocchia, spalle e gomiti. Altezza adulta è stato leggermente ridotto con l’aumento della lordosi lombare. Le articolazioni interfalangee erano prominenti con brevi quinto metacarpo. Come in tutte le famiglie segnalate con mutazioni COL11A2, esame oftalmologico era normale.
ALTRI DISTURBI
Altre malattie con alcune caratteristiche in comune con la sindrome di Stickler includono displasia congenita spondiloepifiseale, 49 displasia di Kniest, 49-51 e la sindrome di Marfan. 52
References
1. ↵
Stickler GB, Belau PG, Farrell FJ, et al.(1965) Hereditary progressive arthro-ophthalmopathy. Mayo Clinic Proc 40:433–455.[Medline][Web of Science]Google Scholar
2. ↵
1. Stickler GB, Pugh DG(1967) Hereditary progressive arthro-ophthalmopathy. II. Additional observations on vertebral abnormalities, a hearing defect, and a report of a similar case. Mayo Clinic Proc 42:495–500.[Web of Science]Google Scholar
3. ↵
1. Temple IK(1989) Stickler’s syndrome. J Med Genet 26:119–126.[FREE Full text]
4. ↵
1. Scott JD(1989) Duke-Elder lecture. Prevention and perspective in retinal detachment. Eye 3:491–515.Google Scholar
5. ↵
1. Seery CM, Pruett RC, Liberfarb RM, Cohen BZ(1990) Distinctive cataract in the Stickler syndrome. Am J Ophthalmol 110:143–148.[Medline][Web of Science]Google Scholar
6. ↵
1. Maumenee IH(1979) Vitreoretinal degeneration as a sign of generalized connective tissue diseases. Am J Ophthalmol 88:432–449.[Medline][Web of Science]Google Scholar
7. ↵
1. Snead MP, Payne SJ, Barton DE, et al.(1994) Stickler syndrome: correlation between vitreoretinal phenotypes and linkage to COL 2A1. Eye 8:609–614.Google Scholar
8. ↵
1. Snead MP, Yates JR, Pope FM, Temple IK,Scott JD.(1996) Masked confirmation of linkage between type 1 congenital vitreous anomaly and COL 2A1 in Stickler syndrome. Graefes Arch Clin Exp Ophthalmol 234:720–721.[CrossRef][Medline][Web of Science]Google Scholar
9. ↵
1. Snead MP(1996) Hereditary vitreopathy. Eye 10:653–663.Google Scholar
10. ↵
1. Nielson CE.(1981) Stickler’s syndrome. Acta Ophthalmol 59:286–295.Google Scholar
11. ↵
1. Cho H,Yamada Y, Yoo TJ.(1991) Ultrastructural changes of cochlea in mice with hereditary chondrodysplasia (cho/cho). Ann N Y Acad Sci 630:259–261.[Medline][Web of Science]Google Scholar
12. ↵
1. Lucarini JW, Liberfarb RM,Eavey RD(1987) Otolaryngological manifestations of the Stickler syndrome. Int J Pediatr Otorhinolaryngol 14:215–222.[CrossRef][Medline][Web of Science]Google Scholar
13. ↵
1. Beighton P(1993) McKusick’s heritable disorders of connective tissue. (Mosby, St Louis), 5th ed..Google Scholar
14. ↵
1. Rai A, Wordsworth P, Coppock JS, Zaphiropoulos GC, Struthers GR.(1994) Hereditary arthro-ophthalmopathy (Stickler syndrome): a diagnosis to consider in familial premature osteoarthritis. Br J Rheumatol 33:1175–1180.[Abstract/FREE Full text]
15. ↵
1. Beals RK.(1977) Hereditary arthro-ophthalmopathy (the Stickler syndrome). Report of a kindred with protrusio acetabuli. Clin Orthop 125:32–35.Google Scholar
16. ↵
1. Liberfarb RM, Goldblatt A.(1986) Prevalence of mitral-valve prolapse in the Stickler syndrome. Am J Med Genet 24:387–392.[CrossRef][Medline][Web of Science]Google Scholar
17. ↵
1. Prockop DJ, Kivirikko KI, Tuderman L, Guzman NA.(1979) The biosynthesis of collagen and its disorders (second of two parts). N Engl J Med 301:77–85.[Medline][Web of Science]Google Scholar
18. ↵
1. Prockop DJ, Kivirikko KI, Tuderman L,Guzman NA.(1979) The biosynthesis of collagen and its disorders (first of two parts). N Engl J Med 301:13–23.[CrossRef][Medline][Web of Science]Google Scholar
19. ↵
1. Francomano CA.(1995) Key role for a minor collagen. Nat Genet 9:6–8.[CrossRef][Medline][Web of Science]Google Scholar
20. ↵
1. Pope FM.(1998) Molecular abnormalities of collagen and connective tissue. Maddison PJ, Isenberg DA, Woo P, Glass DN, editors. Oxford textbook of rheumatology. (Oxford University Press, Oxford), pp 353–404.Google Scholar
21. ↵
1. Prockop DJ, Kivirikko KI.(1995) Collagens: molecular biology, diseases and potentials for therapy. Annu Rev Biochem 64:403–434.[CrossRef][Medline][Web of Science]Google Scholar
22. ↵
1. Upholt WB, Strom CM, Sandell LJ.(1985) Structure of the type II collagen gene. Ann N Y Acad Sci 460:130–140.[CrossRef][Medline][Web of Science]Google Scholar
23. ↵
1. Ballo R, Beighton PH, Ramesar RS.(1998) Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am J Med Genet 80:6–11.[CrossRef][Medline][Web of Science]Google Scholar
24. ↵
1. Horton WA.(1996) Progress in human chondrodysplasias: molecular genetics. Ann N Y Acad Sci 785:150–159.[Medline]Google Scholar
25. ↵
1. Richards AJ, Yates JR, Williams R, et al.(1996) A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet 5:1339–1343.[Abstract/FREE Full text]
26. ↵
1. Martin S, Richards AJ,Yates JRW, Pope FM, Scott JD, Snead MP.(1998) Stickler syndrome types 1 and 2: confirmation of genetic heterogeneity and evidence for another locus. Presentation XXI Meeting Club Jules Gonin, p A34.Google Scholar
27. ↵
1. Mayne R, Brewton RG, Mayne PM, Baker JR.(1993) Isolation and characterization of the chains of type V/type XI collagen present in bovine vitreous. J Biol Chem 268:9381–9386.[Abstract/FREE Full text]
28. ↵
1. Wilkin DJ, Mortier GR, Johnson CL, et al.(1998) Correlation of linkage data with phenotype in eight families with Stickler syndrome. Am J Med Genet 80:121–127.[CrossRef][Medline][Web of Science]Google Scholar
29. ↵
1. Wagner H.(1938) Ein bisher unbekanntes Erbleiden des Auges (Degeneratio hyaloideo-retinalis hereditaria), beobachtet im Kanton Zurich. Klin Monatsbl Augenheilkd 100:840–857.Google Scholar
30. ↵
1. Liberfarb RM, Hirose T, Holmes LB.(1981) The Wagner-Stickler syndrome: a study of 22 families. J Pediatr 99:394–399. [CrossRef][Medline][Web of Science]Google Scholar
31. ↵
1. Brown DM, Graemiger RA, Hergersberg M, et al.(1995) Genetic linkage of Wagner disease and erosive vitreoretinopathy to chromosome 5q13-14. Arch Ophthalmol 113:671–675.[CrossRef][Medline][Web of Science]Google Scholar
32. ↵
1. Fryer AE, Upadhyaya M ,Littler M, et al.(1990) Exclusion of COL2A1 as a candidate gene in a family with Wagner-Stickler syndrome. J Med Genet 27:91–93.[Abstract/FREE Full text]
33. ↵
1. Korkko J, Ritvaniemi P, Haataja L, et al.(1993) Mutation in type II procollagen (COL2A1) that substitutes aspartate for glycine alpha 1-67 and that causes cataracts and retinal detachment: evidence for molecular heterogeneity in the Wagner syndrome and the Stickler syndrome (arthro-ophthalmopathy). Am J Hum Genet 53:55–61.[Medline]Google Scholar
34. ↵
1. Brown DM, Kimura AE, Weingeist TA, Stone EM.(1994) Erosive vitreoretinopathy. A new clinical entity. Ophthalmology 101:694–704.[Medline][Web of Science]Google Scholar
35. ↵
1. Marshall D.(1958) Ectodermal dysplasia. Report of a kindred with ocular deformities and hearing defect. Am J Ophthalmol 45:143–156.[Medline][Web of Science]Google Scholar
36. ↵
1. Shanske AL, Bogdanow A, Shprintzen RJ, Marion RW.(1997) The Marshall syndrome: report of a new family and review of the literature. Am J Med Genet 70:52–57.[CrossRef][Medline]Google Scholar
37. ↵
1. Cohen MM, Jr.(1974) The demise of the Marshall syndrome. J Pediatr 85:878.[Medline]Google Scholar
38. ↵
1. Ayme S, Preus M.(1984) The Marshall and Stickler syndromes: objective rejection of lumping. J Med Genet 21:34–38.[Abstract/FREE Full text]
39. ↵
1. Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH,Warman ML.(1998) Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am J Hum Genet 62:816–823.[CrossRef][Medline][Web of Science]Google Scholar
40. ↵
1. Shanske A, Bogdanow A, Shprintzen RJ, Marion RW.(1998) Marshall syndrome and a defect at the COL11A1 locus. Am J Hum Genet 63:1558–1561.[CrossRef][Medline][Web of Science]Google Scholar
41. ↵
1. Warman ML, Tiller GE, Griffith AJ.(1998) Reply to Shanske et al. Am J Hum Genet 63:1559–1561.[CrossRef][Medline][Web of Science]Google Scholar
42. ↵
1. Weissenbacher G, Zweymuller E.(1964) Coincidental occurrence of Pierre Robin and foetal chondrodysplasia. Monatsschr Kinderheilkd 112:315–317.[Medline]Google Scholar
43. ↵
1. Giedion A, Brandner M, Lecannellier J, et al.(1982) Oto-spondylo-megaepiphyseal dysplasia (OSMED). Helv Paediatr Acta 37:361–380.[Medline]Google Scholar
44. ↵
1. Pihlajamaa T, Prockop DJ, Faber J,et al.(1998) Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher-Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am J Med Genet 80:115–120.[CrossRef][Medline][Web of Science]Google Scholar
45. ↵
1. Brunner HG, van Beersum SE, Warman ML, Olsen BR, Ropers HH, Mariman EC(1994) A Stickler syndrome gene is linked to chromosome 6 near the COL11A2 gene. Hum Mol Genet 3:1561–1564.[Abstract/FREE Full text]
46. ↵
1. Vikkula M, Mariman EC, Lui VC, et al.(1995) Autosomal dominant and recessive osteochondrodysplasias associated with the COL11A2 locus. Cell 80:431–437.[CrossRef][Medline][Web of Science]Google Scholar
47. ↵
1. Sirko-Osadsa DA, Murray MA, Scott JA, Lavery MA, Warman ML, Robin NH.(1998) Stickler syndrome without eye involvement is caused by mutations in COL11A2, the gene encoding the alpha2(XI) chain of type XI collagen. J Pediatr 132:368–371.[CrossRef][Medline][Web of Science]Google Scholar
48. ↵
1. van Steensel MA, Buma P, de Waal Malefijt MC, van den Hoogen FH, Brunner HG.(1997) Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical description of three patients homozygous for a missense mutation in the COL11A2 gene. Am J Med Genet 70:315–323.[CrossRef][Medline][Web of Science]Google Scholar
49. ↵
1. Spranger J, Winterpacht A, Zabel B.(1994) The type II collagenopathies: a spectrum of chondrodysplasias. Eur J Pediatr 153:56–65.[Medline][Web of Science]Google Scholar
50. ↵
1. Kniest W.(1952) Zur abgrenzung der dysostosis enchondrallis von der chondrodystrophie. Z Kinderheilkd 70:633–640.[CrossRef][Medline]Google Scholar
51. ↵
1. Maumenee IH, Traboulsi EI.(1985) The ocular findings in Kniest dysplasia. Am J Ophthalmol 100:155–160.[Medline][Web of Science]Google Scholar
52. ↵
1. Gray JR, Davies SJ.(1996) Marfan syndrome. J Med Genet 33:403–408.[FREE Full text]
53. ↵
1. Brown DM, Vandenburgh K, Kimura AE, Weingeist TA, Sheffield VC, Stone EM.(1995) Novel frameshift mutations in the procollagen 2 gene (COL2A1) associated with Stickler syndrome (hereditary arthro-ophthalmopathy). Hum Mol Genet 4:141–142.[FREE Full text]
54. ↵
1. Ahmad NN, McDonald-McGinn DM, Zackai EH,et al.(1993) A second mutation in the type II procollagen gene (COL2AI) causing Stickler syndrome (arthro-ophthalmopathy) is also a premature termination codon. Am J Hum Genet 52:39–45.[Medline][Web of Science]Google Scholar
55. ↵
1. Williams CJ, Ganguly A, Considine E, et al.(1996) A-2→G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred. Am J Med Genet 63:461–467.[CrossRef][Medline][Web of Science]Google Scholar
56. ↵
1. Ahmad NN, Ala-Kokko L, Knowlton RG, et al.(1991) Stop codon in the procollagen II gene (COL2A1) in a family with the Stickler syndrome (arthro-ophthalmopathy). Proc Natl Acad Sci USA 88:6624–6627.[Abstract/FREE Full text]
57. ↵
1. Brown DM, Nichols BE, Weingeist TA, Sheffield VC, Kimura AE,Stone EM.(1992) Procollagen II gene mutation in Stickler syndrome. Arch Ophthalmol 110:1589–1593.[CrossRef][Medline][Web of Science]Google Scholar
58. ↵
1. Ritvaniemi P, Hyland J, Ignatius J, Kivirikko KI, Prockop DJ, Ala-Kokko L.(1993) A fourth example suggests that premature termination codons in the COL2A1 gene are a common cause of the Stickler syndrome: analysis of the COL2A1 gene by denaturing gradient gel electrophoresis. Genomics 17:218–221.[CrossRef][Medline][Web of Science]Google Scholar
59. ↵
1. Ahmad NN, Dimascio J, Knowlton RG,Tasman WS.(1995) Stickler syndrome. A mutation in the nonhelical 3′ end of type II procollagen gene. Arch Ophthalmol 113:1454–1457.[CrossRef][Medline][Web of Science]Google Scholar
|
|||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Didascalia : |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|