INESTETISMI DELL’ ORECCHIO( Malformazioni congenite ed acquisite)
PADIGLIONE AURICOLARE
Basi
►Definizione: malformazioni congenite o acquisite o difetti del padiglione
auricolare legati ad alterazioni di sviluppo del primo e del secondo arco branchiale, o a traumi, flogosi, resezione di tumori, ustioni, congelamento.
►Epidemiologia: I dati non sono omogenei in quanto c’è poca concordanza sulla metodologia di raccolta dei dati e sulla definizione, Secondo Errocat (1998) in Europa le atresie congenite del cue sono 1 ogni 10000 nati (periodo 1980-1994). con una netta prevalenza delle forme unilaterali (70-85%. Manach. 1987: Schuknecht. 1989: Cremers e Teunissen. 1992) dei maschi e dell’orecchio destro, orecchie ad ansa nel 5% della popolazione, atresie di 1°-3° grado 10-20/100000 neonati/anno (circa 150 nuovi casi per anno in Italia),vedi Fig. 1/a, b).
PADIGLIONE
►Suddivisione dei casi di atresia:
|
|
|
|
|



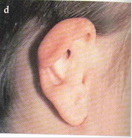
Fig. 1 Orecchio ad ansa in bambino di 10 anni visto dal davanti; b) da dietro; c) appendici preauricolari; d) padiglione ripiegato associato ad atresia del condotto
• Displasie di 1° grado: è presente la maggior parte della struttura anatomica del padiglione (macrotia, coloboma, orecchio ad ansa tipo 1 e 2, piccole anomalie [fistole, cisti, vedi Fig. 1 a-d], Darwin-Höcker, orecchio da satiro)
Definizione chirurgica: la ricostruzione non richiede l’uso di tessuti addizionali
• Displasie di 2° grado: è presente solo una parte delle strutture anatomiche del padiglione (microtia di 2° grado, orecchio ad ansa tipo 3, orecchio piccolo)
Definizione chirurgica: la parziale ricostruzione richiede l’uso addizionale di cute e cartilagine.

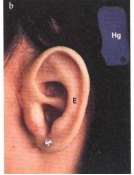

Fig. 1.2 Microtia (talidomide). a, b) Impianto transcutaneo di viti di titanio per fissazione di epitesi (E) e protesi acustica (Hg); c) visto dal davanti.
• Displasie di 3° grado (Microtia di 3° grado, aplasia): non è presente alcuna struttura anatomica di base (per es. atresia auris congenita, sovente associata ad alterazioni di sviluppo del condotto uditivo esterno o dell’orecchio medio, displasia parziale di metà del volto, a distribuzione familiare, da talidomide), vedi Fig. 1.2a-b
Definizione chirurgica: la ricostruzione totale richiede l’uso di cute e notevole quantità di cartilagine
CONDOTTO UDITIVO ESTERNO
Maggiore: atresico
Minore: stenotico
► Clinica; Sintomi: La microtia si può presentare con un ampio spettro di variabilità (Fig. 1). Quando ci si trova di fronte ad una anotia/microtia, vanno tenuti presenti due aspetti: 1. la possibile concomitante malformazione dell’orecchio medio e interno: 2. le possibili malformazioni di altri organi e apparati. In uno studio sulle caratteristiche epidemiologiche della anotia e microtia in California nel decennio 1989-97 (Birth Defects Research Part A 70:472-475. 2004). le malformazioni associate più frequentemente riscontrate sono: quelle oculari (20%), le malformazioni nasali (23.4%), palatoschisi (11,1%), malformazioni muscolo-scheletriche della faccia, mandibola e cranio (23,4%). anomalie delle ossa cranio-facciali (42.4%), anomalie della colonna (14,9%). i difetti cardiaci settali 16.7%, reni/ureteri (6,7%) e genitali maschili (5,1%). conseguenze psicologiche, disturbi funzionali (udito direzionale, difficoltà per occhiali ed apparecchi acustici retroauricolari).
DIAGNOSTICA
►lndispensabile.
• Il primo punto deve essere la definizione morfologica dell’anomalia (Tab. I)
· Anamnesi familiare accurata, anamnesi materna del periodo di gravidanza.
· Ispezione: conformazione (elice, antelice, trago, antitrago e conca normalmente conformati?), angolo tra padiglione auricolare e mastoide.
· Palpazione: elasticità delle cartilagini, presenza di residui cartilaginei
· Sondaggio di fistole o stenosi a livello dell’imbocco del condotto uditivo
esterno,
· Valutazione ORL, esame con microscopio.
· Esami dell’udito: diapason, audiogramma
►Utile in casi particolari:
· Prove vestibolari, ABR per soglia.
· Funzionalità del n. facciale
· Misura delle dimensioni del padiglione.
· Ecografia (anomalie renali e/o altri distretti)
· TAC delle rocche ad alta risoluzione.
· RMN rocche e cerebrale
· RX rachide e/o altri distretti
· Cariotipo
· Genetica molecolare
· Consulto interdisciplinare: pediatra, neurologo, genetista, discussione del caso con i familiari.
· Documentazione fotografica (obbligatoria in caso di intervento).
Tab. I Definizione morfologica dell’anomalia
|
Anomalia simmetrica? Grado della displasia? classificazione |
|
Interessamento delle strutture adiacenti (ossee, nervose, tessuti molli)? |
|
Altre anomalie associate? |
|
Orientamento diagnostico (spettro OAV, sindrome BOR, sdr di Treacher-Collins, altre sindromi) |
DIAGNOSI DIFFERENZIALE
· Padiglione auricolare malformato a seguito di trauma (congelamento, ustione, schiacciamento, ecc).
· Otoematoma organizzato.
· Condrodermatite cronica nodulare dell’elice.
· Tofi gottosi.
· Fistola tubercolare di linfonodo preauricolare.
TERAPIA
►Trattamento, il termine “atresia congenita dell’orecchio” è generalmente usato per descrivere una ampia serie di malformazione dell’orecchio esterno e medio. Anche se il termine atresia implica l’assenza del canale, è usato in senso più ampio anche per indicare da una anomalia lieve con un restringimento del cue, alla assenza completo del canale. Esistono molte divergenze di opinione sulla necessità e opportunità del trattamento; altrettante divergenze sia sulle procedure di scelta sia sui criteri per la valutazione del successo chirurgico.
Vanno innanzi tutto distinti gli interventi in caso di stenosi, da quelli in caso di atresia, Recentemente il successo funzionale delle protesi impiantabili per via ossea (e delle epitesi impiantabili) (Granstrom et al., 1993, 1979) ha certamente ridotto il numero di interventi di chirurgia ricostruttiva del condotto uditivo esterno e li ha spostati in là negli anni, alla seconda adolescenza o addirittura all’età adulta
►Terapia medica: applicazione di epitesi (vedi Fig. 1.2).
►Indicazioni all’intervento:
• Evidente sfigurazione estetica.
• Intolleranza.
• Alterazione psichica.
• Disturbo funzionale secondario (per es. sistemazione di occhiali o di una protesi acustica).
• Padiglione auricolare ad ansa causato da assenza dell’antelice, angolo eccessivamente aperto con la mastoide, assenza del contorno dell’elice.
• Fistole, cisti, appendici cutanee preauricolari.
• Displasia del padiglione, aplasia, microtia, anotia, malformazioni coesistenti del condotto uditivo esterno e dell’orecchio medio.
►Controindicazioni all’intervento:
•Tendenza conosciuta o sospettata alla formazione di cheloidi.
• Stato generale compromesso (valutazione individuale).
• Malattia metabolica (valutazione individuale).
•Infezione locale, flogosi cutanea
►Età alla quale eseguire l’intervento:
• Plastica ricostruttiva del padiglione e displasia di 1° grado di regola tra il 5° ed il 6° anno di vita,
• Displasie di 2° e 3° grado di regola a partire dall’S° anno di vita.
• Quando è previsto un intervento per coesistente atresia del condotto o malformazione congenita dell’orecchio medio, di regola questo deve essere effettuato dopo l’intervento di ricostruzione del padiglione.
►Principi e scopi dell’intervento: eliminare cisti, fistole, appendici cutanee Asportare il tragitto fistoloso dopo riempimento con blu di metilene Escissione di un’appendice cutanea con sutura cutanea immediata.
►Interventi per modificare la forma del padiglione: correzione chirurgica plastica ed estetica corrispondente alla deformità, al fine di ricostruire un padiglione con i suoi contrassegni caratteristici
• orecchi ad ansa: plastica del padiglione (speciale sutura della cartilagine
[per es. Mustardé] evtl, combinata con asportazione di partì cartilaginee
[peres. tecnica di Converse, Pitanguy].
• displasia di I grado: (salvo le “orecchie ad ansa) escissione cartilaginea e
rimodellamento (tecnica di sutura-taglio, taglio-sutura con o senza escis-
ioni di parti molli ed eventuali misure ricostruttive addizionali).
• displasia di II grado: difficile plastica ricostruttiva e delle parti molli in più
tempi con trapianti liberi di cute di preferenza con materiale di sostegno
(vedi avvertenza). Nel caso di difetti parziali del padiglione lembi liberi
compositi dell’orecchio controlaterale .
displasia di III grado : estesa ricostruzione di un padiglione auricolare con
completo scheletro cartilagineo, di preferenza utilizzando cartilagini costali ,autologhe, lembi cutanei e tissutali regionali e liberi, eventualmente con espansione cutanea, di regola in più sedute,
◙Attenzione : per la scelta dei materiali da impiantare tenere conto dello stato attuale della ricerca. Gli impianti omologhi ed eterologhi cartilaginei ed ossei conservati in soluzioni di Cialit e mertiolato non sono più impiegabili allo stato attuale delle conoscenze; lo stesso per la cartilagine prelevata da animali .Il materiale alloplastico ha un tasso di estrusione più elevato rispetto al materiale proprio dell’organismo. Grazie a tecniche di” tissue engineering”, si spera sia in futuro possibile coltivare in vitro una cartilagine auricolare della forma desiderata, da impiantare successivamente sottocute ed una volta rivestitita da cute, trasferirla nella sede definitiva con chirurgia microvascolare.
Collocazione di epitesi: sono possibili tecniche conservative (colla) e preferibilmente chirurgiche (ancoraggio osseo) e queste ultime si rivelano particolarmente importanti anche se problematiche nei bambini (vedi Fig. 1-2a-c in combinazione con protesi acustica).
Ambulatorialmente/con ricovero:
L’asportazione di piccole fistole, cisti ed appendici cutanee può essere eseguita, ambulatoriamente. Plastiche ricostruttive del padiglione e interventi per displasia di I grado possono essere effettuati ambulatoriamente o con ricovero
secondo l’età del paziente, l’assistenza a casa e l’estensione dell’intervento.
• Tutti gli altri interventi ricostruttivi con ricovero.
Prognosi:
•orecchi ad ansa: un reintervento è desiderato dai pazienti nel 15% dei casi.
•displasia di II e III grado , anotia: un reintervento è desiderato dal paziente o è necessario (più sedute) nella maggioranza dei casi,
Diagnosi. Data la possibilità di concomitanti malformazioni in altri organi e apparati microtia/atresia non isolata ma sindromica). l’approccio a questa patologia deve essere multidisciplinare (Neonatologo. Genetista. Audiologo. ORL. Neuroradiologo. Chirurgo maxillofacciale. Chirurgo plastico. Oculista. Neurochirurgo). I l primo punto deve essere la definizione morfologica dell’anomalia (Tab. I)
Otoplastica/ricostruzione del padiglione auricolare
Padiglione auricolare abdotto (orecchio ad ansa), difetti del padiglione auricolare, malformazioni
Principi generali
Tutti gli interventi vanno condotti sotto copertura antibiotica (vedi sotto).
La tecnica operatoria è descritta prendendo come esempio l’intervento di plastica del padiglione auricolare (plastica dell’antelice, vedi sotto).
Principi in caso di difetti del padiglione: ricostruzione con lembi cutanei peduncolati dalle sedi vicine (regione retroauricolare) o con grafts compositi (ad es. prelevando materiale dal padiglione auricolare sano).
Principi per la ricostruzione del padiglione auricolare: allestimento di lembi cutanei dalle regioni circostanti, creazione di lembi tubulati, loro trasposizione per la creazione del contorno del padiglione, successivo impianto sottostante di cartilagine (in più sedute). Oppure: impianto di uno scheletro” di ne auricolare sagomato da cartilagine costale sotto la cute al di sopra c mastoideo dopo espansione cutanea, oppure sotto la cute del collo, con successivamente trasposizione dell’innesto peduncolato nella sede definitiva.Anestesia
Anestesia locale (adulti).Anestesia generale (bambini).
Tecnica operatoria, esempio: plastica dell’ antelice ,otopessia
Marcatura della struttura dell’antelice mediante infissione di aghi in corrispondenza della superficie esterna.
Escissione cutanea retroauricolare a losanga nella zona demarcata. La cute’ va scollata fino al margine dell’elice e in direzione della mastoide,
Escissione di tessuto connettivo e muscolare dal piano mastoideo,
Lungo la marcatura vengono praticate, con la punta dì un bisturi o con fresa di diamante, 2-3 incisioni cuneiformi a decorso parallelo comprende pericondrio e cartilagine.
Con la sutura del pericondrio (materiale di sutura incolore 4/O), la cartilagine assume la conformazione desiderata
Eventualmente vengono praticati 2-3 ulteriori punti di sutura tra perb del padiglione auricolare e periostio del piano mastoideo (pessia) cutanea, medicazione compressiva.
Suggerimenti
Ambulatoriale/con ricovero:
• Intervento dopo i 5 anni di età
Generalmente intervento ambulatoriale.
Trattamento postoperatorio:
• Cambio della medicazione nel 1 giorno dopo l’intervento (controllo C toma, raccolta di siero, necrosi cutanea da compressione), Rimozione punti in 1O giornata.
· In caso di dolore, soprattutto se pulsante, medicazione immediata e trollo delle condizioni della ferita (ematoma).
· Fasciatura auricolare durante la notte per 3 settimane
Microtia e atresia auris congenita
Definizione. La microtia è una malformazione congenita caratterizzata da un ridotto sviluppo dell’orecchio esterno, che varia da un padiglione più piccolo, alla totale assenza. Nei casi più gravi sono presenti solo residui embrionari: l’assenza del padiglione viene definita anotia, La microtia è spesso associata a stenosi o atresia del condotto uditivo estrn. Entrambe queste anomalie possono essere associate con altre anomalie dell’orecchio. più frequentemente dell’orecchio medio, meno frequentemente dell’orecchio interno. Va tenuto presente che esiste una stretta correlazione tra grado di malformazione dell’orecchio esterno e frequenza e gravità delle malformazioni dell’orecchio medio. Al contrario non esiste una correlazione tra grado di malformazione dell’orecchio esterno e frequenza e gravità delle malformazioni dell’orecchio interno: le malformazioni dell’orecchio interno sono riferite più frequenti nelle embriopatie tossiche. virali e genetiche. Tra le malformazioni dell’orecchio interno, non vi sono apparenti spiegazioni della maggior frequenza della ipoplasia del canale semicircolare laterale.
Epidemiologia I dati non sono omogenei in quanto c’è poca concordanza sulla metodologia di raccolta dei dati e sulla definizione, Secondo Errocat (1998) in Europa le atresie congenite del cue sono 1 ogni 10000 nati (periodo 1980-1994). con una netta prevalenza delle forme unilaterali (70-85%. Manach. 1987: Schuknecht. 1989: Cremers e Teunissen. 1992) dei maschi e dell’orecchio destro,
Clinica. La microtia si può presentare con un ampio spettro di variabilità (Fig. 1). Quando ci si trova di fronte ad una anotialmicrotia. vanno tenuti presenti due aspetti: 1. la possibile concomitante malformazione dell’orecchio medio e interno: 2. le possibili malformazioni di alul organi e apparati. In uno studio sulle caratteristiche epidemiologiche della anotia e microtia in California nel decennio 1989-97 (Birth Defects Research Part A 70:472-475. 2004). le malformazioni associate più frequentemente riscontrate sono: quelle oculari (20%), le malformazioni nasali (23.4%), palatoschisi (11,1%), malformazioni muscolo-scheletriche della faccia. mandibola e cranio (23,4%). anomalie delle ossa cranio-facciali (42.4%), anomalie della colonna (14,9%). i difetti cardiaci settali (16.7%). reni/ureteri (6,7%) e genitali maschili (5,1%).
|
|
Fig I Ampio spettro variabilità della microtia (A. Martini e G,P Garani).
|

Diagnosi. Data la possibilità di concomitanti malformazioni in altri organi e apparati microtia/atresia non isolata ma sindromica). l’approccio a questa patologia deve essere multidisciplinare (Neonatologo. Genetista. Audiologo. ORL. Neuroradiologo. Chirurgo maxillofacciale. Chirurgo plastico. Oculista. Neurochirurgo). Il primo punto deve essere la definizione morfologica dell’anomalia (Tab. I)
|
Anomalia simmetrica? Grado della displasia? classificazione |
|
Interessamento delle strutture adiacenti (ossee, nervose, tessuti molli)?
|
|
Altre anomalie associate? |
|
Orientamento diagnostico (spettro OAV, sindrome BOR, sdr di Treacher-Collins, altre sindromi)
|
Tab. I Definizione morfologica dell’anomalia
APPROFONDIMENTO
Classificazioni Molte sono le classificazioni che si trovano in letteratura Recentemente, l’European Group on Genetics of Hearing lmpairment (HEAR) ha proposto un sistema di classificazione delle anomalie congenite di tipo sistematico, dall’orecchio esterno alla corteccia cerebrale ECEAI European Congenital Ear Anomly Inventory (v Tab.) L’ECEAI ha lo scopo di fornire una descrizione anatomica sistematica e dettagliata delle varie componenti dell’orecchio non si base su una classificazione eziologica, ma mette in risalto la necessità di valutare tutte le differenti parti del sistema uditivo
|
|
|
PADIGLIONE Definizione generale: sono riconoscibili la maggior parte delle strutture del padiglione Definizione chirurgica: la ricostruzione non richiede l’uso di tessuti addizionali Definizione generale: sono riconoscibili solo alcune strutture del padiglione Definizione chirurgica: la parziale ricostruzione richiede l’uso addizionale di cute e cartilagine Definizione generale: non è riconoscibile alcuna struttura dell’orecchio esterno Definizione chirurgica: la ricostruzione totale richiede l’uso di cute e notevole quantità di cartilagine CONDOTTO UDITIVO ESTERNO Maggiore: atresico Minore: stenotico
|
|
ORECCHIO MEDIO |
|
CATENA OSSICULARE Tipo 1: anchilosi congenita della staffa* Fissità della platina con struttura normale o monopodale Tipo 2: anchilosi congenita della staffa più altra anomalia della catena ossiculare* Fissità timpanica (manico del martello e/o processo lungo) Tipo 3: anomalia congenita della catena ossiculare con platina mobile Fissità timpanica (manico del martello e/o processo lungo) Persistenza dell’arteria stapediale |
|
ORECCHIO INTERNO |
|
A COCLEA MALFORMATA O ASSENTE Aplasia completa del labirinto (deformità di Michel) Aplasia cocleare: assenza della coclea, canali semicircolari e vestibolo normali o malformati lpoplasia cocleare: dotto cocleare piccolo, canali semicircolari e vestibolo normali o malformati
|
|
COCLEA NORMALE Displasia del canale semicircolare laterale e del vestibolo: vestibolo dilatato e cs.I. dilatano e corto; gli altri canali semicircolari sono normali Acquedotto vestibolare allargato, canali semicircolari normali, vestibolo dilatato o normale
|
|
CONDOTTO UDITIVO INTERNO E SISTEMA UDITIVO CENTRALE Non esiste una classificazione universalmente accettata
|
|
assenza del tendine dello stapedio
|
Tab ECEAI sintesi delle anomalie principali.
Quindi vanno predisposte le indagini utili all’inquadramento del quadro clinico (Tab. II). l’orientamento attuale è quello di eseguire in un periodo “precoce” (entro i 6 mesi) anche le indagini neuroradiologiche, non solo per venire incontro “all’ansia” dei genitori. ma perché un inquadramento diagnostico corretto è essenziale per evitare errori di impostazione terapeutica.
Trattamento, ill termine “atresia congenita dell’orecchio” è generalmente usato per descrivere una pia serie di malformazione dell’orecchio esterno e medio. Anche se il termine atresia implica l’assenza del canale, è usato in senso più ampio anche per indicare da una anomalia lieve con un restringimento del cue, alla assenza completo del canale. Esistono molte divergenze di opinione sulla necessità e opportunità del trattamento; altrettante divergenze sia sulle procedure di scelta sia sui criteri per la valutazione del successo chirurgico.
Vanno innanzi tutto distinti gli interventi in caso di stenosi, da quelli in caso di atresia, Recentemente il successo funzionale delle protesi impiantabili per via ossea (e delle epitesi impiantabili) (Granstrom et al., 1993, 1979) ha certamente ridotto il numero di interventi di chirurgia ricostruttiva del condotto uditivo esterno e li ha spostati in là negli anni, alla seconda adolescenza o addirittura all’età adulta (Fig. 2 e 3),
|
Valutazione audiologica con ABR per soglia
|
|
Ecografia (anomalie renali e/o altri distretti)
|
|
TC rocche |
|
RMN rocche e cerebrale |
|
RX rachide e/o altri distretti |
|
Cariotipo
|
|
Genetica molecolare |
Tab. II. Indagini in caso di microtia/atresia congenita.
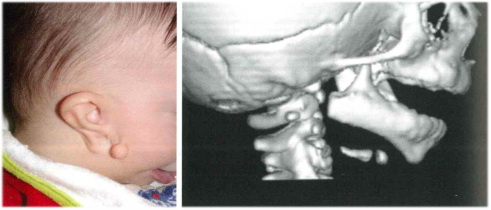
Fig 2. Quadro clinico e radiologico in un caso di sindrome di Goldenhar. È evidente a
destra la microtia di III grado, la atresia del CUE, la presenza di appendice preauricolare e l’atresia del cue alla TAC.
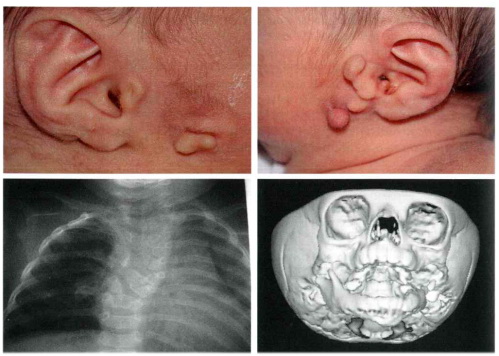
Fig 3 Caso di OAV con appendici preauricolari ed auricolari bilaterali e alla Rx
cranio-rachide presenza di emispondili a livello dorsale
BIBLIOGRAFIA
|
|
Lambert P.R., Dodson E.E. Congenital malformations of the external auditory canal Otolaryngol. Clin. North Am. 1996 ; 29 : 741-760
Goeringer G.C. Development of the ear Pediatric otology and neurotology New York: Lippincott-Raven Press (1998). 3-10
Marsh K.L., Dixon M.J. Treacher Collins syndrome Adv. Otorhinolaryngol. 2000 ; 56 : 53-59 [cross-ref]
Treacher Collins Syndrome Collaborative Group Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome Nat. Genet. 1996 ; 12 : 130-136
Stoll C., Viville B., Treisser A., Gasser B. A family with dominant oculoauriculovertebral spectrum Am. J. Med. Genet. 1998 ; 78 : 345-349 [cross-ref]
Tasse C., Majewski F., Bohringer S., Fischer S., Ludecke H.J., Gillessen-Kaesbach G., e al. A family with autosomal dominant oculo-auriculo-vertebral spectrum Clin. Dysmorphol. 2007 ; 16 : 1-7 [cross-ref]
Kallen B., Mastroiacovo P., Robert E. Major congenital malformations in Down syndrome Am. J. Med. Genet. 1996 ; 65 : 160-166 [cross-ref]
Harris J., Kallen B., Robert E. The epidemiology of anotia and microtia J. Med. Genet. 1996 ; 33 : 809-813 [cross-ref]
Beahm E.K., Walton R.L. Auricular reconstruction for microtia: part I. Anatomy, embryology, and clinical evaluation Plast. Reconstr. Surg. 2002 ; 109 : 2473-2482 [cross-ref]
Manac’h Y. L’aplasie de l’oreille. Des principes au cas particulier Arch. Pediatr. 1995 ; 2 : 413-414
Francois M., Wiener-Vacher S.R., Falala M., Narcy P. Audiological assessment of infants and children with preauricular tags Audiology 1995 ; 34 : 1-5 [cross-ref]
Meurman Y. Congenital microtia and meatal atresia; observations and aspects of treatment Arch. Otolaryngol. 1957 ; 66 : 443-463
Nagata S. A new method of total reconstruction of the auricle for microtia Plast. Reconstr. Surg. 1993 ; 92 : 187-201 [cross-ref
Cohen M.M., Rollnick B.R., Kaye C.I. Oculo-auriculo-vertebral spectrum: an updated critique Cleft Palate J. 1989 ; 26 : 276-286
Ohtani I., Dubois C.N. Aural abnormalities in Klippel-Feil syndrome Am. J. Otol. 1985 ; 6 : 468-471
Valdez B.C., Henning D., So R.B., Dixon J.Dixon M.J. The Treacher-Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor Proc. Natl. Acad. Sci. USA 2004 ; 101 : 10709-10714 [cross-ref]
Herrmann B.W., Karzon R., Molter D.W. Otologic and audiologic features of Nager acrofacial dysostosis Int. J. Pediatr. Otorhinolaryngol. 2005 ; 69 : 1053-1059 [cross-ref]
O’Callaghan M., Young I.D. The Townes-Brocks syndrome J. Med. Genet. 1990 ; 27 : 457-461
Chang E.H., Menezes M., Meyer N.C., Cucci R.A., Vervoort V.S., Schwartz C.E., e al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences Hum. Mutat. 2004 ; 23 : 582-589 [cross-ref]
Dyce O., McDonald-McGinn D., Kirschner R.E., Zackai E., Young K., Jacobs I.N. Otolaryngologic manifestations of the 22q11.2 deletion syndrome Arch. Otolaryngol. Head Neck Surg. 2002 ; 128 : 1408-1412
Kountakis S.E., Helidonis E., Jahrsdoerfer R.A. Microtia grade as an indicator of middle ear development in aural atresia Arch. Otolaryngol. Head Neck Surg. 1995 ; 121 : 885-886
Ishimoto S., Ito K., Karino S., Takegoshi H., Kaga K., Yamasoba T. Hearing levels in patients with microtia: correlation with temporal bone malformation Laryngoscope 2007 ; 117 : 461-465 [cross-ref]
Teunissen E.B., Cremers W.R. Classification of congenital middle ear anomalies. Report on 144 ears Ann. Otol. Rhinol. Laryngol. 1993 ; 102 : 606-612
Teunissen E.B., Cremers W.R. Classification of congenital middle ear anomalies. Report on 144 ears Ann. Otol. Rhinol. Laryngol. 1993 ; 102 : 606-612
Brown D.J., Kim T.B., Petty E.M., Downs C.A., Martin D.M., Strouse P.J., e al. Autosomal dominant stapes ankylosis with broad thumbs and toes, hyperopia, and skeletal anomalies is caused by heterozygous nonsense and frameshift mutations in NOG, the Gene encoding noggin Am. J. Hum. Genet. 2002 ; 71 : 618-624 [cross-ref]
Chandrasekhar S.S., De la Cruz A., Garrido E. Surgery of congenital aural atresia Am. J. Otol. 1995 ; 16 : 713-717
Jahrsdoerfer R.A., Yeakley J.W., Aguilar E.A., Cole R.R., Gray L.C. Grading system for the selection of patients with congenital aural atresia Am. J. Otol. 1992 ; 13 : 6-12
Carvalho G.J., Song C.S., Vargervik K., Lalwani A.K. Auditory and facial nerve dysfunction in patients with hemifacial microsomia Arch. Otolaryngol. Head Neck Surg. 1999 ; 125 : 209-212
Roman S., Moncla A., Philip N., Triglia J.M. Les surdités mixtes de l’enfant Rapport de la Société Française d’ORL, Génétique et maladies ORL : (2005).
Swartz J.D., Faeber E.N. Congenital malformations of the external and middle ear: high resolution CT findings of surgical import AJR Am. J. Roentgenol. 1985 ; 144 : 501-506
Takegoshi H., Kaga K., Chihara Y. Facial canal anatomy in patients with mandibulofacial dysostosis: comparison with respect to the severities of microtia and middle ear deformity Otol. Neurotol. 2005 ; 26 : 803-808 [cross-ref]
Yamane H., Takayama M., Sunami K., Tochino R., Morinaka M. Disregard of cholesteatoma in congenital aural stenosis Acta Otolaryngol. 2007 ; 127 : 221-224 [cross-ref]
Takegoshi H., Kaga K., Chihara Y. Facial canal anatomy in patients with mandibulofacial dysostosis: comparison with respect to the severities of microtia and middle ear deformity Otol. Neurotol. 2005 ; 26 : 803-808 [cross-ref]
Jahrsdoerfer R.A., Gillenwater A.M. The facial nerve in congenital ear malformations Eur. Arch. Otorhinolaryngol. 1994 ; S299-S301
Linstrom C.J., Meiteles L.Z. Facial nerve monitoring in surgery for congenital auricular atresia Laryngoscope 1993 ; 103 : 406-415
Patel N., Shelton C. The surgical learning curve in aural atresia surgery Laryngoscope 2007 ; 117 : 67-73 [cross-ref]
Jahrsdoerfer R.A. Transposition of the facial nerve in congenital aural atresia Am. J. Otol. 1995 ; 16 : 290-294
Han D., Zhao S., Wang D., Guo J., Dai H. Vestibulotomy above a severely displaced facial nerve Acta Otolaryngol. 2005 ; 125 : 962-965 [cross-ref]
Lambert P.R. Congenital absence of the oval window Laryngoscope 1990 ; 100 : 37-40
Siegert R., Mattheis S., Kasic J. Fully implantable hearing aids in patients with congenital auricular atresia Laryngoscope 2007 ; 117 : 336-340 [cross-ref]
McKinnon B.J., Jahrsdoerfer R.A. Congenital auricular atresia: update on options for intervention and timing of repair Otolaryngol. Clin. North Am. 2002 ; 35 : 877-890 [cross-ref]
Albert S., Roger G., Rouillon I., Chauvin P., Denoyelle F., Derbez R., e al. Congenital stapes ankylosis: study of 28 cases and surgical results Laryngoscope 2006 ; 116 : 1153-1157 [cross-ref]
Welling D.B., Merrell J.A., Merz M., Dodson E.E. Predictive factors in pediatric stapedectomy Laryngoscope 2003 ; 113 : 1515-1519 [cross-ref